
高效液相色谱法测定乌头汤有效成分含量的方法建立
2023-02-17 03:12:04王佳郑淑晶李阳胡德冷向阳王淑敏
特产研究 2023年1期
王佳,郑淑晶,李阳,胡德,冷向阳,王淑敏
(长春中医药大学,吉林 长春 130117)
乌头汤最早始载于张仲景的《金匮要略——中风历节病脉证并治第五》中,“病历节不可屈伸疼痛,乌头汤主之”,临床应用已有千年之久[1]。该方由川乌(制)、麻黄、白芍、黄芪和甘草(炙)组成,现代临床应用主要用于治疗膝骨关节炎、类风湿性关节炎、腰椎骨关节炎、腰椎间盘性疼痛及肩关节周围炎效果显著[2-4],且副作用小。
有效成分是中药材及中药复方的物质基础,是研究的重中之重[5]。利用现代科学技术对乌头汤中各味药材的化学成分及药理作用进行研究发现,制川乌中主要活性成分为生物碱类,而单酯型生物碱如苯甲酰新乌头原碱、苯甲酰次乌头原碱和苯甲酰乌头原碱为其主要活性物质,具有消炎镇痛的功效[6]。麻黄碱和伪麻黄碱为麻黄的主要活性成分,已有研究表明其具有良好的抗炎、镇痛及免疫调节等功效[7],毛蕊异黄酮葡萄糖苷是黄芪中黄酮类的主要成分,具有抗炎、抗病毒和免疫调节作用,可使氢化可的松致免疫功能低下模型鼠的细胞免疫功能恢复至正常水平。甘草中的甘草酸和甘草苷也具有相关药理作用[8]。此外,毛霞等[9]通过分子对接技术、药代动力学及SPR技术相结合,确立了芍药苷为乌头汤中有效成分之一。上述成分均为乌头汤治疗关节炎等相关疾病的有效成分,因此,本实验针对乌头汤中已有确切药效的化合物为其有效成分,对其进行综合评价,建立高效液相色谱分析方法,为乌头汤的质量控制、多指标成分含量测定以及今后提取工艺和安全用药提供研究方法和科学依据。
1 仪器与材料
1.1 试药
芍药苷(18032901)、甘草酸(18060805)、苯甲酰乌头原碱(18110710)、苯甲酰次乌头原碱(18032807)、苯甲酰新乌头原碱(18032406)、新乌头碱(17111010)、次乌头碱(17111310)、乌头碱(17110910)、毛蕊异黄酮葡萄糖苷(18031920)、甘草苷(18032801)购自于北京世纪奥科生物技术有限公司,盐酸麻黄碱(171241-201809)、盐酸伪麻黄碱(171237-201510)购自于中国食品药品检定研究院,乙腈、甲醇为色谱纯(美国赛默飞世尔科技有限公司)、娃哈哈纯净水(杭州娃哈哈有限公司),磷酸等其他试剂均为分析纯。
1.2 仪器与设备
岛津LC-20AT系统(SPD-M20A PDA检测器、SIL-20A自动进样器,日本岛津科技有限公司),电子天平(MS105DU,梅特勒-托利多仪器有限公司),超声波清洗器(AS3120A,天津奥特赛恩斯仪器有限公司),冷冻干燥仪(Epsilon2-4Lsplus),高速离心机(TGL16M,湖南凯达科学仪器有限公司),pH计(PHSJ-3F,上海仪电科学仪器股份有限公司)。
1.3 药材
制川乌、黄芪、麻黄、白芍和炙甘草经长春中医药大学王淑敏教授鉴定分别为毛茛科植物乌头(Aconitum carmichaelii Debx)的干燥母根,豆科黄芪属植物蒙古黄芪[Astragalus membranaceus(Fish.)Bge.var.mongholicus(Bge.)Hsiao]的干燥根,麻黄科植物草麻黄(Ephedra sinica Stapff)的干燥草质茎,毛茛科植物芍药(Paeonia lactiflora Pall)的干燥根,豆科植物甘草(Glycyrrhiza uralensis Fisch)干燥根的蜜制品。以上药材均购自吉林省宏检大药房。
2 实验方法
2.1 色谱条件
色 谱 柱:Waters XSelect CSH C18(4.6 mm 250mm,5m),流动相:10 mmol/L磷酸二氢铵,磷酸调pH 2.1(A)-乙腈(B),采用梯度洗脱:0~8 min,95.5%A;8~11 min,95.5%~91%A;11~16 min,91%~82%A;16~30 min,82%~80% A;30~35 min,80%~77% A;35~43 min,77%~72% A;43~51 min,72%~57% A;51~59min,57%~5%A;59~65min,5%A,流速:1mL/min,柱温:35℃,检测波长:210 nm、235 nm,进样量:5L。
2.2 对照品溶液配制
精密称取适量盐酸麻黄碱、盐酸伪麻黄碱、芍药苷、毛蕊异黄酮葡萄糖苷、甘草苷、苯甲酰新乌头原碱、苯甲酰乌头原碱、苯甲酰次乌头原碱、新乌头碱、乌头碱、次乌头碱和甘草酸于容量瓶内,甲醇溶解定容至刻度,配制成浓度分别为0.46 mg/mL、0.32 mg/mL、0.53mg/mL、0.17 mg/mL、0.50 mg/mL、0.90 mg/mL、0.96 mg/mL、0.88 mg/mL、0.40 mg/mL、0.45 mg/mL、0.31mg/mL和0.60mg/mL的标准混合对照品溶液备用。
2.3 供试品溶液配制
称取原处方4份(麻黄36 g、黄芪36 g、炙甘草36 g、白芍36 g、制川乌24 g)于煎药壶内,加入12倍量水浸泡30 min,武火煮沸,文火慢煎90 min,提取2次,合并提取液,进行冷冻干燥,即得乌头汤提取物冻干粉。
2.4 阴性对照液配制
分别取不含有上述各味药材的复方4份,按2.3项下进行制备,即得各组阴性对照溶液。
2.5 样品溶液前处理
取乌头汤冻干粉0.2 g,加95%的甲醇5 mL,超声提取30 min,于10 000 r/min,离心10 min取上清液,即得供试品溶液1;另取乌头汤冻干粉2g,加入1 mol/L的盐酸1 mL,10 mL乙醚超声提取15 min,弃去乙醚液,再向残渣中加入1 mL氨试液,10 mL乙醚萃取2次,合并萃取液,30℃氮气吹干,1mL0.5%盐酸甲醇定容,即得供试品溶液2。
3 实验结果
3.1 方法学考察
3.1.1 专属性考察取上述对照品溶液,阴性对照品溶液按2.1项下色谱条件进行检测,结果见图1和图2。结果显示各成分之间均无干扰,专属性良好。

图1 12种标准品HPLC色谱图Fig.1 HPLC chromatograms of twelve standard substances

图2 乌头汤阴性对照HPLC色谱图Fig.2 HPLC chromatogram of the wutou decoction negative control
3.1.2线性关系考察称取上述对照品标准溶液0.1mL、0.5mL、1mL、2mL、5mL、8mL和10mL于10mL容量瓶内,甲醇定容至刻度,配制成不同浓度的混合标准品溶液,进样量5L,以各标准品峰面积为y轴,浓度为x轴绘制标准曲线,结果见表1,表明各成分在相应浓度范围内线性良好。
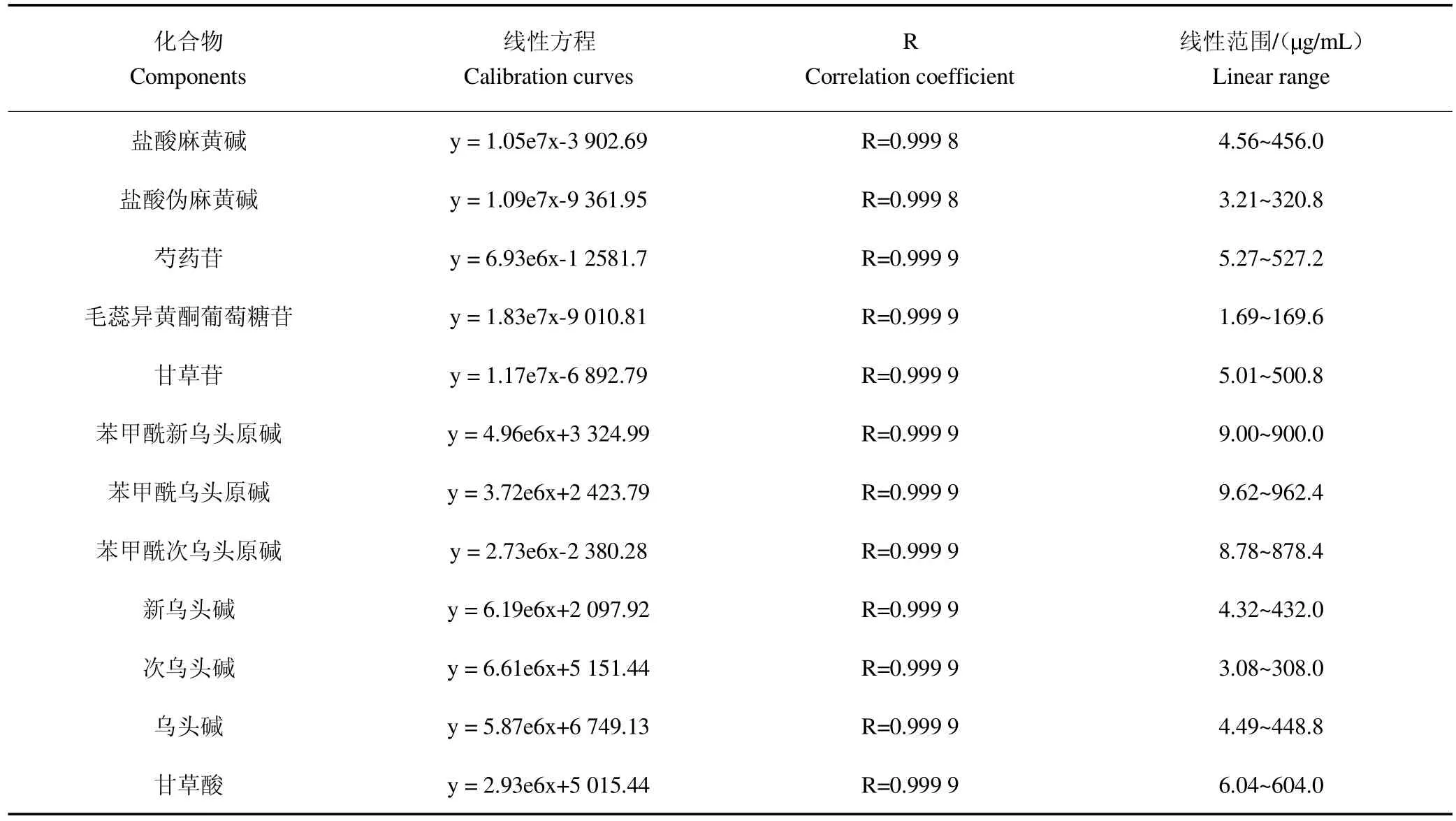
表1 12种化合物线性方程及线性范围Table 1 The calibration curves of twelve compounds
3.1.3 精密度实验取混合对照品溶液,按上述2.1色谱条件连续进样6次。计算各成分峰面积的RSD,考察仪器精密度。结果显示盐酸麻黄碱、盐酸伪麻黄碱、芍药苷、毛蕊异黄酮葡萄糖苷、甘草苷、苯甲酰新乌头原碱、苯甲酰乌头原碱、苯甲酰次乌头原碱、新乌头碱、乌头碱、次乌头碱和甘草酸的RSD分别为0.32%、0.25%、0.46%、0.58%、0.36%、0.39%、0.42%、0.61%、0.52%、0.58%、0.46%和0.41%,表明该仪器精密度良好。
3.1.4 稳定性实验精密量取同一供试品溶液,密闭置于室温条件下,分别于0 h、2 h、4 h、8 h、10 h和24 h按2.1色谱条件进行测定,结果显示盐酸麻黄碱、盐酸伪麻黄碱、芍药苷、毛蕊异黄酮葡萄糖苷、甘草苷、苯甲酰新乌头原碱、苯甲酰乌头原碱、苯甲酰次乌头原碱、新乌头碱、乌头碱、次乌头碱和甘草酸的RSD分别为2.56%、3.23%、2.21%、1.68%、2.06%、2.87%、3.26%、1.78%、2.32%、3.01%、2.75%和2.48%,表明供试品溶液放置24 h基本稳定。
3.1.5 重复性实验精密称取同一批供试品,按2.3项下的方法平行制备6份供试品溶液,并按2.1项下色谱条件进行检测,结果显示盐酸麻黄碱、盐酸伪麻黄碱、芍药苷、毛蕊异黄酮葡萄糖苷、甘草苷、苯甲酰新乌头原碱、苯甲酰乌头原碱、苯甲酰次乌头原碱、新乌头碱、乌头碱、次乌头碱和甘草酸的RSD分别为1.96%、1.69%、1.56%、1.78%、2.06%、2.36%、3.04%、2.41%、1.89%、2.96%、1.78%和2.76%,表明该方法重复性良好。
3.1.6 加样回收实验精密量取0.2 g已知含量的供试品6份,各组分按1:1的比例分别精密加入一定量混合对照品,按2.3项下方法进行供试品制备,并按2.1项下色谱条件进行检测。结果显示盐酸麻黄碱、盐酸伪麻黄碱、芍药苷、毛蕊异黄酮葡萄糖苷、甘草苷、苯甲酰新乌头原碱、苯甲酰乌头原碱、苯甲酰次乌头原碱、新乌头碱、乌头碱、次乌头碱和甘草酸的平均回收率在90%~108%之间,RSD小于5%,表明各成分回收率良好。
3.2 含量测定
按照2.3项下方法制备3批供试品溶液,并按2.1项下方法进行检测,计算每批样品中各成分的含量,结果见表2。
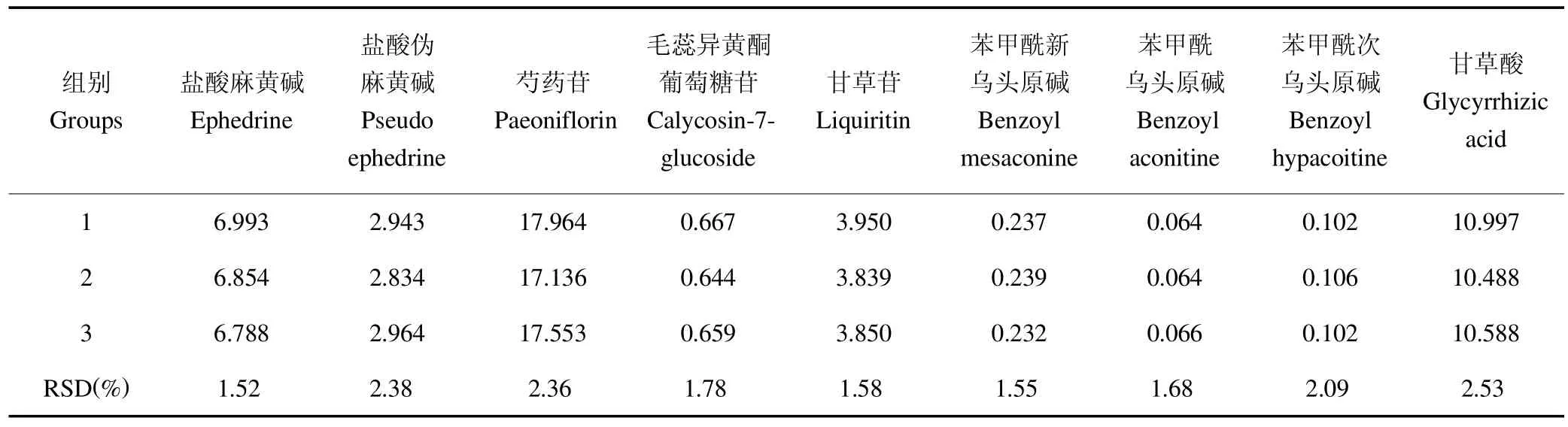
表2 含量测定结果Table 2 The results of content determination mg/g
4 讨论
现代医学研究表明,乌头汤成分复杂,具有多成分、多靶点的治疗特点,现有分析方法无法采用同一条件对乌头汤中各类成分进行统一有效地定量分析,而是根据不同化合物类型采用不同条件和方法进行测定[10,11],其原因主要是由于组成乌头汤的5味药材基源各不相同,复方中有效成分包含生物碱类、黄酮类和萜类等多种结构类型,进而导致色谱分离过程中存在较大难度。尤其是生物碱类化合物,在液相分离过程中,往往伴随着峰形拖尾和载样量低等情况,导致色谱峰宽较宽,峰高较矮,色谱峰的分离不利以及灵敏度差等问题[12]。因此针对上述问题,本实验开发一种高效液相色谱方法,用于乌头汤有效成分的含量测定。
该方法首次将乌头汤中不同结构类型的有效成分进行统一测定。通过对流动相pH及缓冲盐进行筛选发现,当流动相pH呈酸性时,各成分峰形良好。而采用磷酸盐作为缓冲盐溶剂时,相较于醋酸铵等有机盐,低波长下,紫外吸收明显减弱,背景噪音影响最小,基线更为平顺,进而有助于提高灵敏度。此外对比不同厂家、不同类型的色谱柱,最终确定Waters Xseclect C18作为本方法的色谱柱,其特点在于固定相表面具有杂化带电颗粒,可与碱性化合物溶质间产生静电排斥,阻止生物碱类化合物向硅醇基靠近,从而改善峰形拖尾[13],进而避免向流动相中加入扫尾剂(如三乙胺)或使用碱性流动相等条件,影响其他类型化合物稳定性或降低色谱柱耐用性,而导致的检测结果不稳定等问题[14]。优化上述条件并结合梯度洗脱,最终确立了本方法各项参数。与现有技术相比,该方法高效,省时省力,成本低廉,适用于科学研究及生产实践,且稳定可靠,各成分间分离度良好,且与阴性对照液对比分析,无假阳性及干扰峰存在。此外该方法也同时将乌头中容易引起中毒反应的毒性成分双酯型乌头碱(新乌头碱、乌头碱、次乌头碱)[15]一并纳入检测范围,在对乌头汤中9种有效成分进行定量分析的同时,也对3种毒性成分进行监测,确保在使用乌头过程中,由于炮制不善等其他因素所导致的中毒反应,提高用药安全性。
由于乌头汤中成分复杂,各有效成分含量差异较大,因此本实验采用两种前处理方法进行样品制备。方法1针对于麻黄碱、伪麻黄碱、芍药苷、毛蕊异黄酮葡萄糖苷、甘草酸及甘草苷等含量较高的成分。方法2则对于乌头类生物碱进行样品的除杂与富集[10],通过酸化成盐提高其水溶性的同时,乙醚萃取去除脂溶性杂质成分,进一步碱化使乌头类生物得以游离,进而增加其在乙醚中的溶解性,提高萃取效率。采用该方法对工艺验证样品中的双酯型生物碱进行测定,低于人体毒性反应的最低剂量0.2 mg[16],符合用药安全。经方法学考察验证,该方法专属性强,精密度、稳定性和重复性RSD均小于3%。加样回收实验中,各指标成分回收率在90%~108%,符合药典相关规定,可为今后乌头汤的质量标准研究和新剂型开发提供科学依据。
猜你喜欢
中国饲料(2021年17期)2021-11-02 08:15:14
理化检验-化学分册(2020年5期)2020-06-15 11:36:12
中成药(2018年11期)2018-11-24 02:57:36
中成药(2018年2期)2018-05-09 07:19:50
广东饲料(2016年5期)2016-12-01 03:43:22
中成药(2016年4期)2016-05-17 06:07:56
合成化学(2015年10期)2016-01-17 08:56:37
应用化工(2014年1期)2014-08-16 13:34:08
中国药业(2014年17期)2014-05-26 09:07:41
发明与创新·大科技(2014年3期)2014-04-29 07:38:59
