
氮硫共掺杂多孔碳材料制备及其电容性能研究
2023-02-17 07:21雷舒霖杨思莉彭付康付旭东
电源技术 2023年1期
雷舒霖,杨思莉,彭付康,彭 倩,付旭东
(湖北工业大学材料与化学工程学院绿色轻工材料湖北省重点实验室,湖北武汉 430068)
超级电容器与普通二次电池相比有较高的功率密度,与传统电容器相比有较高的能量密度[1]。根据电极材料不同,超级电容器可分为双电层电容器和赝电容器。双电层电容器的电极材料为活性碳[2]、碳纳米管[3]和石墨烯[4]等碳材料。赝电容器的电极材料有金属氧化物[5]、导电聚合物[6]等。相比于赝电容材料,活性碳具有电化学稳定性好、成本低和资源丰富等优势,但是其比电容较低。杂原子(氮、磷、硫等)掺杂可提高活性碳的比电容,因为活性碳上杂原子位点可以吸附更多离子[7]。目前大多通过热处理含有杂原子的碳前驱体来制备杂原子掺杂的活性碳,但是在热处理过程中,前驱体会分解并挥发,导致活性碳中杂原子掺杂量低[8]。
本文通过在聚对苯乙烯磺酸(PSS)水溶液中原位聚合苯胺,构筑三维网状结构聚苯胺(PANI)/PSS 水凝胶,冷冻干燥后得到碳材料前驱体。PSS 中磺酸根基团和PANI 中胺基存在静电交联作用(图1),能够减缓热处理过程中氮、硫基团的分解挥发,从而提高碳材料中氮和硫的掺杂量。通过优化PANI和PSS 的比例,制得碳材料的比电容达到160 F/g (@1 A/g),且具有优异的循环稳定性。

图1 聚苯胺和聚对苯乙烯磺酸之间的静电交联作用示意图
1 实验
1.1 样品的制备
(1)纯PANI 的制备
量取5 mL 去离子水倒入烧杯中,然后加入0.493 1 g 苯胺单体(An),将其放在超声波清洗器中超声10 min,同时以玻璃棒进行搅拌使其混合均匀,作为溶液A。再称取1.164 g 过硫酸铵溶于5 mL 蒸馏水中作为溶液B。将溶液A 和溶液B 分别冷却至4 ℃,再混合均匀于4 ℃环境中静置12 h 使苯胺聚合。
(2)不同浓度的PSS/PANI 制备
量取5 mL 去离子水后倒入烧杯中,然后加入0.493 1 g 单体An 和0.458 0 g PSS,将其放在超声清洗器中超声10 min,同时以玻璃棒进行搅拌使其混合均匀,作为溶液C(其中An 的浓度约为0.5 mol/L,PSS 的结构单元的浓度约为0.25 mol/L)。称取1.164 g 过硫酸铵溶于5 mL 蒸馏水中作为溶液D。溶液C和溶液D 冷却至4 ℃之后,将两溶液混合均匀于4 ℃环境中静置12 h 使苯胺聚合。苯胺单体浓度固定,改变PSS 的结构单元浓度以制备不同样品。
(3)多孔碳材料制备
将上述制备的不同的PANI/PSS 样品及纯PANI 放入冷冻干燥箱中进行干燥,干燥时间为24 h。干燥后研磨成粉末,称取一定质量的样品置于石英方舟中,再置于管式反应炉中,在升温以前在管式反应炉内抽真空然后通氮气,循环3 次以上以排除炉内空气。设定程序开始升温,先以10 ℃/min 的升温速率升温至300 ℃,然后以5 ℃/min 的升温速率升温至800 ℃,在氮气的保护下,保持800 ℃碳化2 h。随后降温至室温,另外3 组不同碳化温度(600、700、900 ℃)的样品与此类似。表1 为7 个样品的统计。
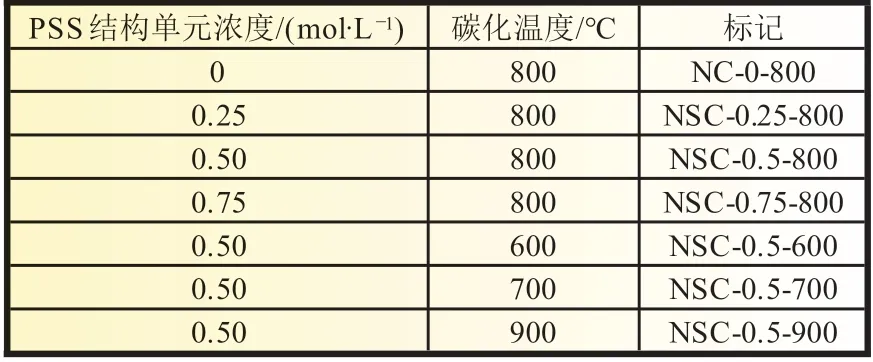
表1 样品制备统计(An 浓度固定为0.5 mol/L)
1.2 样品表征
采用扫描电子显微镜(SEM,SU8010,Hitachi)表征碳材料形貌。用X 射线光电子能谱(XPS,PHI 5000 VerseProbe III,ULVAC PHI)分析碳材料中C、N、O、S 四种元素的含量。对样品的比表面积和孔结构进行了表征(BET,PLUS HD88,Micromeritics),通过BET 方程计算所测碳材料样品的比表面积与孔隙分布。用傅里叶变换红外光谱(FTIR,TENSOR II,Bruker)研究碳材料前驱体的分子结构。
1.3 电极制备及测试
称取80 mg 所制备的多孔碳材料、10 mg 乙炔黑和10 mg PTFE 粘合剂放置在研钵中,滴加异丙醇进行研磨。研磨均匀后取出于平板上压成薄膜,冲压成直径约1 cm 的圆片,并在60 ℃的真空烘箱中干燥6 h。干燥后,电极在2.5 MPa 的压力下在镍泡沫集流体上冷压120 s。于6 mol/L 的KOH 水溶液中用三电极系统进行电化学测试。三电极体系是以Pt 片作为对电极,Hg/HgO 电极(电解质溶液1 mol/L KOH 溶液)为参比电极,所得的样品为工作电极。在电化学工作站(CHI760E,上海辰华)进行循环伏安(CV)测试和电化学阻抗谱(EIS)测定,其中CV 扫描的电压窗口为-0.9~0.1 V,扫速为100 mV/s,EIS 在开路条件下测试,扰动电压幅值为5 mV,频率从10 kHz 到0.1 Hz。恒流充放电(GCD)采用蓝电电池测试系统(CT2001A,武汉蓝电)进行测试,充放电之前电池静置半小时。
CV 和GCD 计算比电容公式如下:

式中:I(A)为电流密度,A/g;∫I(A)dV为CV 曲线积分面积;V为CV 曲线的电压窗口,V;ν为CV 曲线扫描速度,V/s;m为电极中活性材料质量,g;i为放电电流密度,A/g;t为放电时间,s;U为放电时的电压降,V。
2 结果与讨论
2.1 聚苯胺的合成情况
图2 为PANI 和PANI/PSS 红外谱图,PANI 在1 563、1 490、1 304、1 175 和825 cm-1处显示了特征峰,其中1 563 和1 490 cm-1处峰为醌环和苯环中的C-C 伸缩振动,1 304 和1 175 cm-1处峰为C-N 和C=N 的伸缩振动,825 cm-1处峰为C-H 的面外弯曲振动,表明PANI 被成功合成[9]。PANI/PSS 在1 031 和1 004 cm-1处有吸收峰,为-SO3-的对称振动[10],且PANI 在1 304 cm-1处峰移动至1 293 cm-1处,表明PSS 的-SO3-与PANI 的C-N 存在静电交联作用[6]。
2.2 碳化温度对多孔碳材料性能的影响
如图3(a)所示,不同碳化温度样品的CV 曲线都呈现矩形,随着碳化温度的不断提高,氮、硫共掺杂多孔碳电极的电流密度逐渐升高,但当碳化温度提高至900 ℃时,其电流密度又略微降低。图3(b)是四种碳材料的GCD 曲线,均为对称的三角形,随着碳化温度的提高,碳材料的放电比电容依次为110、120、160、154 F/g。CV 和GCD 的结果表明碳化温度为800 ℃时,氮、硫共掺杂的多孔碳材料具有最高的比电容。
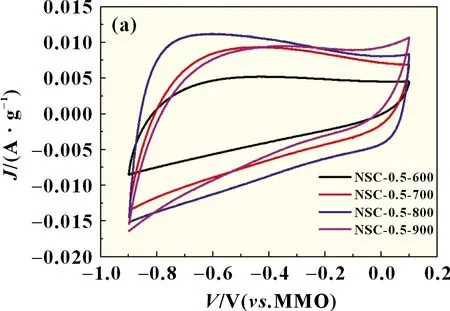

图3 样品在100 mV/s扫速下的循环伏安曲线(a)和1 A/g电流密度下的恒流充放电曲线(b)
2.3 氮、硫共掺杂对多孔碳材料性能的影响
图4(a)~(c)为NSC-0.5-800 在不同放大倍率下的SEM 图片,显示样品呈现相互连接的网状多孔结构,部分保留了前驱体的结构。为了进一步得到氮、硫共掺杂碳材料的孔结构特点,进行氮气吸脱附实验来测定比表面积和孔结构,图4(d)为NC-0-800、NSC-0.25-800、NSC-0.5-800 和NSC-0.75-800 的氮气吸脱附曲线,均为I 型曲线的特性,即在低压区吸附量增幅很大,表明具有大量存在的介孔结构,并随着介孔结构进一步的吸附而饱和时,出现吸脱附曲线中的平台效应,因此从曲线形状可以判断出所制备的碳材料以介孔为主。样品NC-0-800、NSC-0.25-800、NSC-0.5-800 和NSC-0.75-800 的BET 比表面积分别为989、1 170、1 127 和658 m2/g,随着PSS浓度提高,BET 比表面积呈现先上升后下降的趋势,对于NSC-0.75-800,由于过高的PSS 浓度导致苯胺聚合困难[8],因此比表面积急剧下降。

图4 SEM 形貌和氮气吸脱附曲线
从图5 的X 射线光电子能谱(XPS)图中可以看出,所制备的样品可以清楚地观察到C 1s、O 1s、N 1s 和S 2p 特征峰,各种元素的含量如表2 所示,NSC-0.25-800、NSC-0.5-800 和NSC-0.75-800 中的氮、硫元素掺杂量都高于NC-0-800 的,表明PSS 和PANI 的静电交联作用能够提高氮、硫元素掺杂量。当An 的浓度和PSS 的结构单元浓度相等时(NSC-0.5-800),氮、硫元素掺杂量最高,与文献相比(表3),NSC-0.5-800 的杂元素掺杂量处于较高水平。NC-0-800 存在微量的S 元素,这是引发剂过硫酸铵的残留导致的。

图5 NC-0-800(a)和NSC-0.5-800(b)的XPS 全谱图

表2 不同PSS 浓度样品的N、S、C、O 元素掺杂含量 %(原子分数)
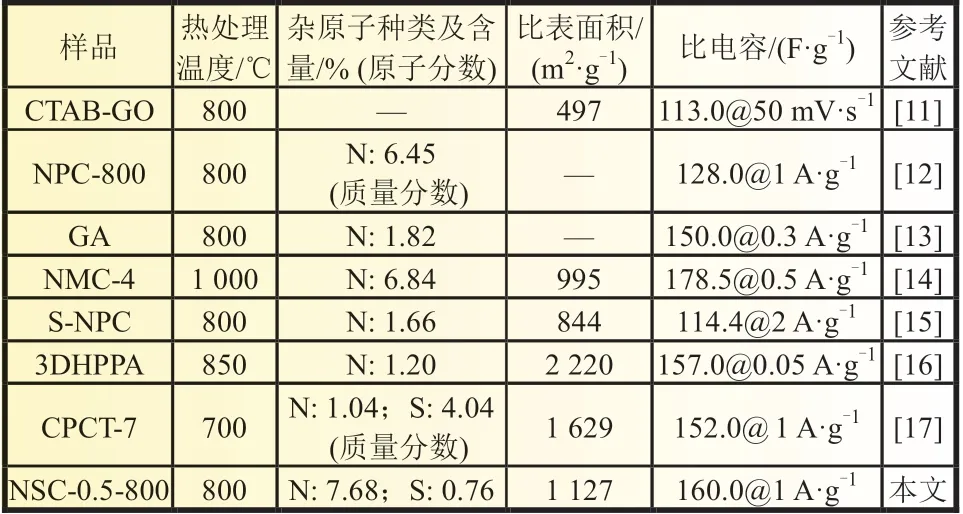
表3 NSC-0.5-800 样品参数与文献对比
NC-0-800、NSC-0.25-800、NSC-0.5-800、NSC-0.75-800 在6 mol/L KOH 溶液中,-0.9~0.1 V 扫描电压窗口内,100 mV/s扫速下的CV 曲线如图6(a)所示。NC-0-800 样品的CV 曲线有轻微扭曲,随着PSS 结构单元浓度的提高,CV 曲线逐渐变为矩形。图6(b)为样品在1 A/g 电流密度下的GCD 曲线,可以观察到对称三角形状,这是典型的理想超级电容器GCD 曲线。通过计算可以得出,NC-0-800、NSC-0.25-800、NSC-0.5-800、NSC-0.75-800 的比电容分别为110、152、160、150 F/g,和杂原子(氮和硫)掺杂量的变化趋势一致,其中NSC-0.5-800中N和S的总掺杂量最高,比电容也最大,和文献相比(表3),NSC-0.5-800 的比电容处于较高水平。NSC-0.75-800 样品由于过高的PSS浓度导致苯胺聚合困难[8],虽然能增加硫元素掺杂量,但是氮元素掺杂量反而降低,所以比电容低于NSC-0.5-800。
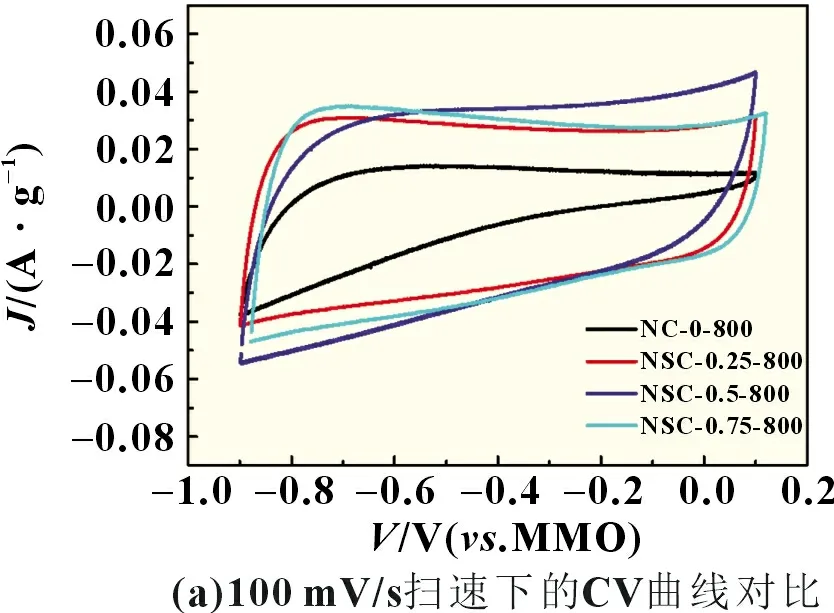

图6 循环伏安曲线和恒流充放电曲线
图7(a)为电化学阻抗Nyquist 对比曲线图,图中曲线与实轴的交点可得欧姆阻抗,NC-0-800、NSC-0.25-800、NSC-0.5-800 和NSC-0.75-800的欧姆阻抗分别为0.24、0.40、0.45 和0.48 Ω,其中NC-0-800 的阻抗最小,表明杂原子掺杂会增加欧姆阻抗。图7(b)为NSC-0.5-800 恒流充放电的循环稳定性图,在电流密度为1 A/g 时,NSC-0.5-800 的比电容为160 F/g。在经过5 000 次充放电循环后,比电容升高为168 F/g,可能随着循环次数的增加,电极材料与电解液接触更充分,氮、硫共掺杂碳材料拥有良好的循环稳定性。

图7 电化学阻抗曲线和循环稳定性曲线
3 结论
本文利用PANI 和PSS 的静电相互作用制备碳材料前驱体,通过改变PSS 的含量来调节碳材料中氮、硫原子在碳材料里的掺杂量,碳材料最佳的制备条件为:An 的浓度和PSS 的结构单元浓度相等(都为0.5 mol/L),碳化温度为800 ℃;结合SEM、BET、XPS 和FTIR 等物理化学表征分析对材料的表面结构及杂原子掺杂影响进行了详细的研究,测试表明PSS 和PANI 的静电交联作用可以提高碳材料的氮、硫元素掺杂量,且NSC-0.5-800 的比表面积(1 127.22 m²/g)高于NC-0-800 的(989.30 m²/g)。当电流密度为1 A/g 时,NSC-0.5-800 的比电容为160 F/g,相比于NC-0-800 的110 F/g 提高了25.6%。同时,NSC-0.5-800 具有优异的循环稳定性。
猜你喜欢
环境卫生工程(2021年4期)2021-10-13
物理之友(2020年12期)2020-07-16
电子制作(2019年22期)2020-01-14
山东冶金(2019年5期)2019-11-16
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年2期)2017-01-20
广西大学学报(自然科学版)(2016年6期)2017-01-04
通信电源技术(2016年6期)2016-04-20
华北理工大学学报(社会科学版)(2015年3期)2016-01-11
