
基于E2F靶点基因集和免疫亚型差异的肝细胞癌预后风险评分模型的建立
2023-02-17 02:46何锶赵杨朱永乾吴卓翼吴英谢君蓉郑登烨简红梅
中国普通外科杂志 2023年1期
何锶,赵杨,朱永乾,吴卓翼,吴英,谢君蓉,郑登烨,简红梅
(中国人民解放军陆军军医大学基础医学院 1. 学员三大队十一队 2. 学员四大队十二队 3. 学员三大队九队,重庆 400038;4. 中国人民解放军陆军军医大学第一附属医院 肝胆外科, 重庆 400038)
原发性肝癌是目前我国第4位常见恶性肿瘤及第2位肿瘤致死病因,严重威胁我国人民的生命和健康[1]。其中原发性肝细胞癌(hepatocellular carcinoma,HCC)是肝癌最常见的类型,发生率占原发性肝癌的75%~85%[2]。尽管近年来原发性HCC的临床治疗模式不断发展,肝癌患者的预后生存情况仍然较差,患者5年生存率仅18%左右[3],预后预测仍然面临巨大挑战。预后风险评分模型可以预测个体在将来是否发生某种结局。近年来,预测模型研究呈明显上升趋势,在肿瘤领域,如乳腺癌、前列腺腺癌、肺腺癌等中已有诸多应用[4-6]。目前,在HCC领域,已有基于基因的风险评分模型被成功建立以预测患者预后[7],但大多只是大范围地筛选基因,没有聚焦在某一已知与癌症发生相关的基因集上,这会为进一步深入的机制研究带来困难。因此,有目的地筛选HCC治疗的关键基因及相关通路,建立新的基因预后模型,预测HCC的潜在治疗靶点具有重大意义和可行性。
转录因子E2F相关基因在调节肿瘤进展,参与血管生成促进肿瘤转移等各方面起着重要作用[8]。另一方面免疫疗法最近受到广泛关注,越来越多的证据[9]表明免疫相关基因可预测肝癌预后。近年来,许多研究已证实E2F基因家族和免疫微环境相关的基因标志物是癌症的重要预后因素,E2F-1基因缺失可导致PTEN丢失诱导的锯齿状肿瘤的发生率和恶化程度增加[10],17个免疫相关基因建立的分类系统能有效预测早期肺鳞状细胞癌患者的预后[11],又如E2F2、3、6、8的高表达与胰腺癌患者的肿瘤分期有关且E2F基因的表达与免疫浸润显著相关[12]。因此本研究旨在利用TCGA数据库开发一种新的E2F基因家族和免疫微环境相关的基因标志物,建立风险评分模型以预测HCC患者的预后并找到治疗靶点。
1 资料与方法
1.1 数据来源与数据预处理
本研究从TCGA数据库(https://portal.gdc.cancer.gov)中下载了424例Liver Hepatocellular Carcinoma(LIHC)患者的临床特征以及60 484个mRNA的表达数据集。从分子特征数据库(MSIGDB)中下载共50组hallmark基因集(癌症特征基因集合),该基因集合表示了明确的生物状态或过程,下载免疫标志基因集,该基因集可表示免疫系统内的细胞状态和调动。基于上述详细数据,将424例LIHC患者根据TCGA数据库的样本编号原则分为有癌组(374例)和无癌组(50例)。此外,还记录了以下临床信息:性别、年龄、肿瘤大小、淋巴结转移数、远处器官转移情况、UICC分期和BMI。最后,对261例有明确生存时间且生存时间不为零的患者进行了分类。一般临床特征详见表1。
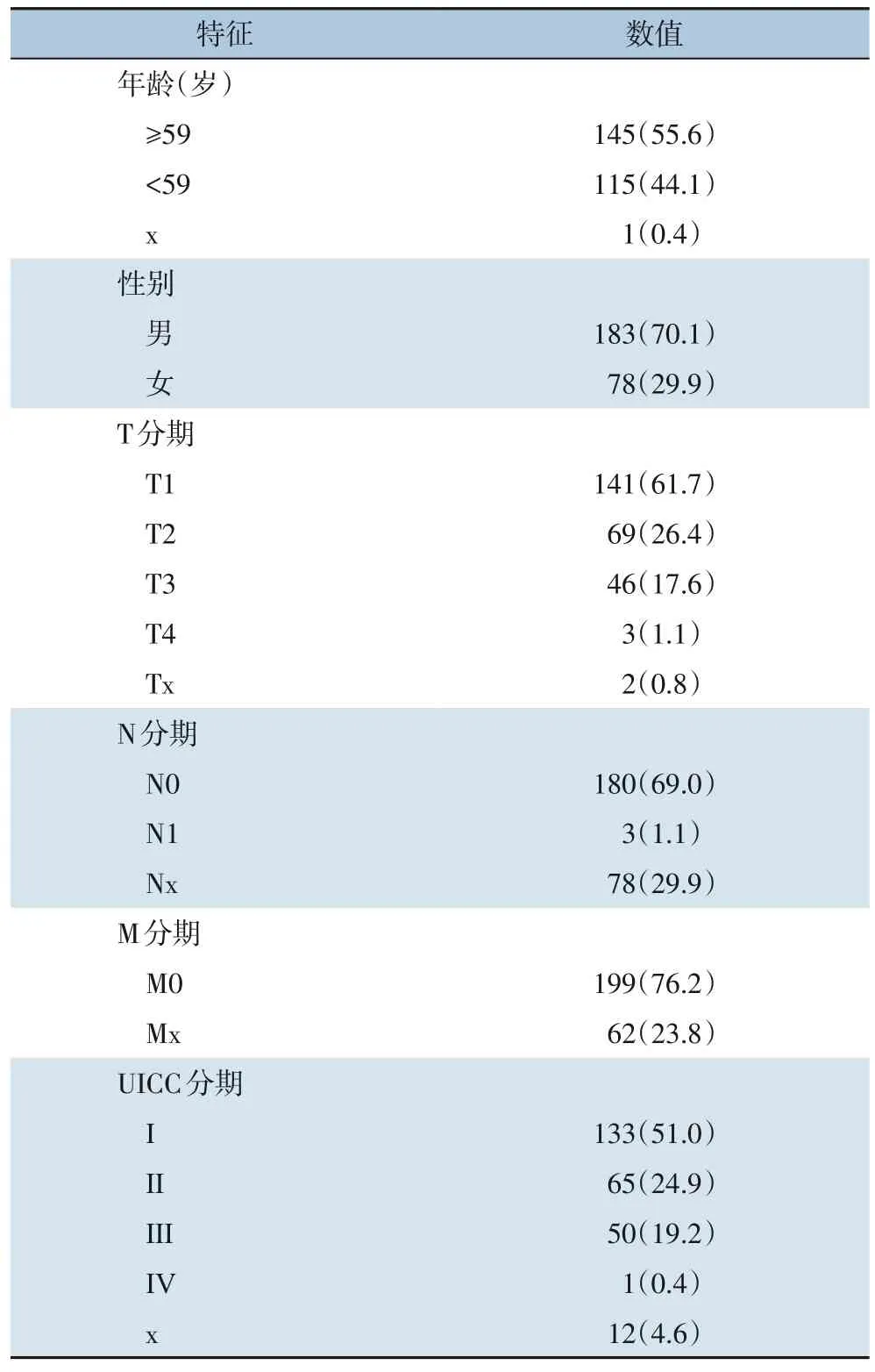
表1 本研究中HCC患者的临床特征[n(%)]Table 1 Clinical characteristics of HCC patients in this study [n (%)]
1.2 基因集富集分析与基因集单样本富集分析
使用“DESeq2”包进行基因表达量的差异分析得到9 149个差异基因(P<0.05),进一步使用“clusterProfiler”包对这9 149个差异基因进行GSEA基因富集分析,以识别在有癌组和无癌组中存在显著统计学差异且富集程度最高的基因组用于进一步研究。基因集单样本富集分析(ssGSEA)是通过扩展GESA实现的,使用签名中的基因和其余基因的经验累积分布函数生成富集分数[13]。用“GSVA”包对374例有癌组样本进行基因集单样本富集分析,可得到各样本在免疫系统内不同细胞状态和调动下的富集得分,再使用“hclust”包对富集得分聚类得到两种免疫相关亚型,最后使用R包“DESeq2”对两组免疫亚型样本的mRNA表达量进行差异分析得到存在显著统计学差异的免疫亚型差异基因(P<0.05)。
1.3 构建预后特征模型
使用glmnet包,采用Cox法进行10倍似然交叉验证,纳入LIHC中的261例有明确生存时间且生存时间不等于零的样本,分别对富集分析中富集得分最高的基因集的基因和免疫亚型差异基因通过Lasso回归进行变量筛选。同样使用glmnet包,采用Cox法进行十倍似然交叉验证,将两者筛选后的基因一起基于表达水平与Lasso回归分析得出的系数加权的线性组合,建立了预后风险评分模型。风险评分=基因1的表达×β1+基因2的表达×β2+…+基因n×βn的表达,并绘制可用于临床评分的列线图。
1.4 预后模型的评估与验证
使用“ROCR”包对风险预测模型对截止时间内生存死亡结局的预测能力通过受试者工作特征曲线(ROC)及曲线下面积(AUC)值进行检验,以验证模型预测生存状态和生存时间的敏感度和特异度。以中位风险评分为分界点,通过中位值将261例患者分为高风险和低风险亚组。使用“survival”包和“survminer”包,绘制Kaplan-Meier生存曲线并进行Log-rank检验。相同地,对BMI值、淋巴转移、远处转移、肿瘤大小和UICC分期的常见临床指标绘制Kaplan-Meier生存曲线并进行Log-rank检验,与风险预测模型相比较。将通过Log-rank检验的各项指标进行单因素和多因素Cox回归分析,以检查风险评分是否是可用数据范围内的一个独立预后因素。在考虑不同UICC分期亚型时,本研究还绘制了I、Ⅱ分期和Ⅲ、Ⅳ分期两亚型的高、低风险的Kaplan-Meier生存曲线,确立风险评分模型的适用范围。随后,本研究检测了有癌组和无癌组之间构成模型的基因的差异表达和基因改变情况,通过蛋白互作网络寻找模型中的关键基因以求发现新的可能药物靶点。372个TCGA HCC样本的构成风险模型的基因的改变量从Cbioportal数据库(http://www.cbioportal.org)查询,蛋白互作信息从String数据库(https://cn.string-db.org)中下载并在Cytoscape软件中分析可视化。
1.5 统计学处理
所有统计分析均使用R软件(版本4.1.0,https://www.r-project.org)进行,P<0.05为差异有统计学意义。整体流程图如图1。
图1 整体流程图Figure 1 Overall flow chart
2 结 果
2.1 富集得分最高的E2F靶点及其相关基因
在差异基因中,E2F靶点基因组的200个基因富集程度高,归一化后的富集得分最高,标准化P值为1.00E-10和多重假设检验矫正后的P值(q值)为6.25E-10(图2A-B)。Lasso回归分析筛选得到两个基因:SSRP1,KIF2C(图2C-D)。
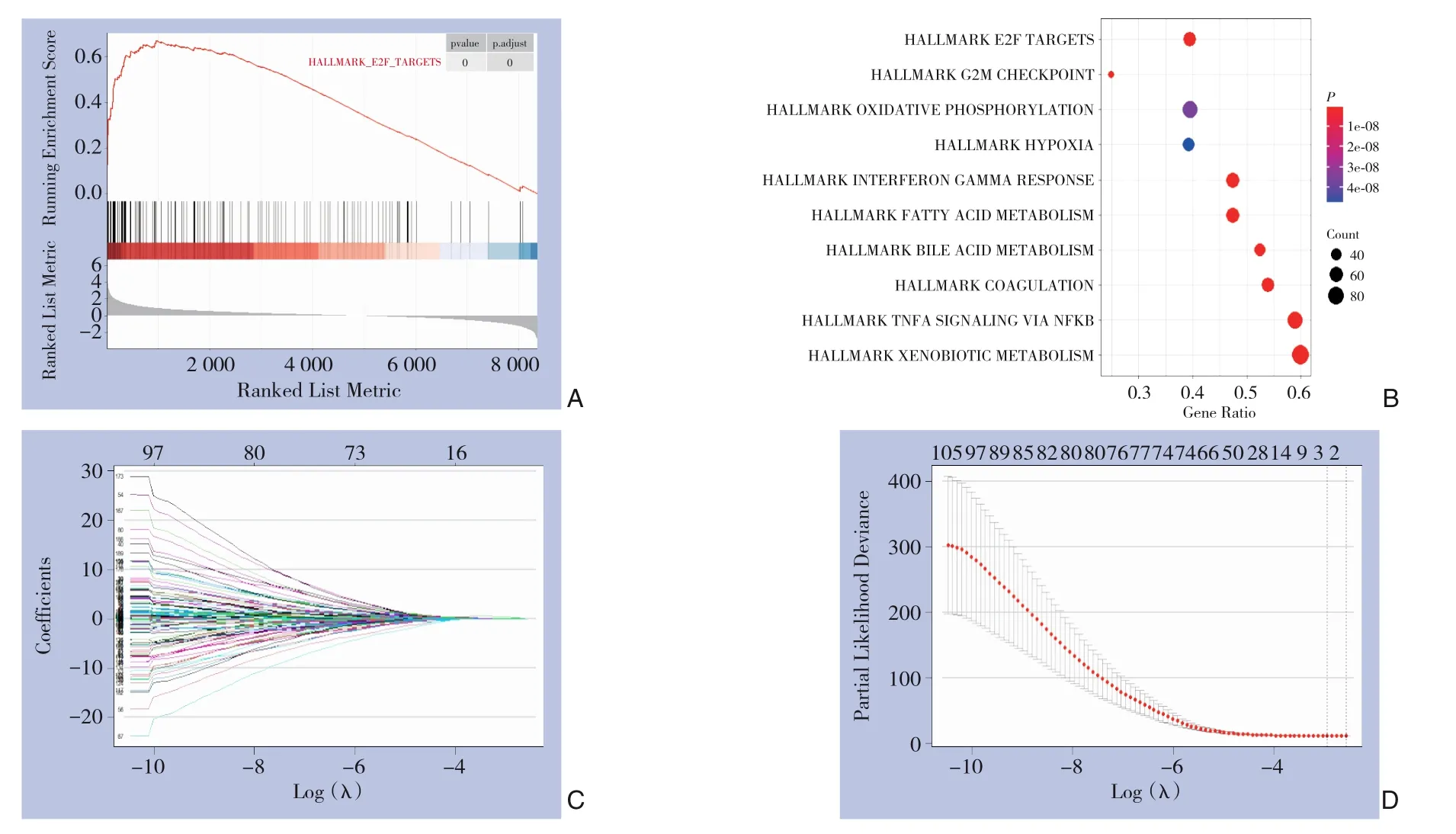
图2 富集得分最高的E2F靶点及其相关基因的鉴定 A:区分有、无癌组的E2F靶点基因集富集图;B:按归一化后的富集得分排序的10个标志基因集的P值及基因集包含基因数量;C:所选特征的系数由λ参数表示;D:偏似然偏差和对数(λ)用Lasso-Cox回归模型绘制Figure 2 E2F targets with the highest enrichment score and identification of related genes A: Enrichment plots of E2F target gene sets differentiated between in cancer and non-cancer group; B: The P-value and the number of genes of top 10 marker gene sets with the highest normalized enrichment score; C: The coefficients of the selected features represented by the λ parameter; D: Partial likelihood deviation and logarithm (λ) constructed with the Lasso-Cox regression model
2.2 免疫亚型及免疫相关差异表达基因
根据样本在免疫系统内不同细胞状态和调动下的富集得分进行聚类,将样本分成了富集得分差异明显的两组(图3A)。两组免疫亚型样本差异存在统计学意义的基因(P<0.05)有242个,其中23个下调,219个上调(图3B)。Lasso回归分析筛选得到6个基因,即CYR61,FBLN5,LPA,SAA1,SDC3,SERPINE1(图3C-D)。
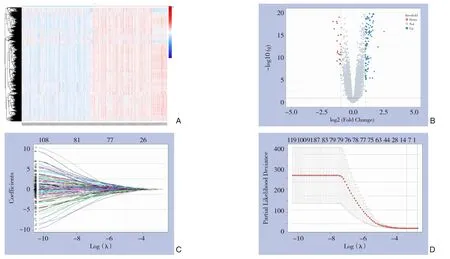
图3 免疫亚型及免疫相关差异表达基因的鉴定 A:有癌组样本按富集得分的聚类分组;B:两免疫亚组间差异表达mRNA的火山图;C:所选特征的系数由λ参数表示;D:偏似然偏差和对数(λ)用Lasso-Cox回归模型绘制Figure 3 Immune subtypes and identification of immune-related differentially expressed genes A: The samples in the cancer group distinguished according to the enrichment score; B: Volcano map of differentially expressed mRNAs between two immune subgroups; C: The coefficients of the selected features represented by the λ parameter; D: Partial likelihood deviation and logarithm (λ) constructed with the Lasso-Cox regression model
2.3 构建预后模型
将E2F靶点基因集筛选得到的两个基因和免疫亚型差异筛选得到的6个基因共同进行Lasso回归,完成变量筛选和预后模型建立,得到7-mRNA预后模型:风险评分=-0.55×CYR61表达-0.18×FBLN5表 达-0.17×LPA表 达-0.06×SAA1表 达+0.31×SDC3表 达+0.38×SERPINE1表 达+1.08×SSRP1表达(图4A)。7个基因通过等比例风险检验(P>0.1),对生存风险的作用在不同时间点变化的比例基本一致。还构建了多因素Cox回归模型,做出列线图(图4B)。对该7-mRNA预后模型对截止时间内生存死亡结局的预测能力进行ROC分析,AUC值为0.846(图4C-D)。
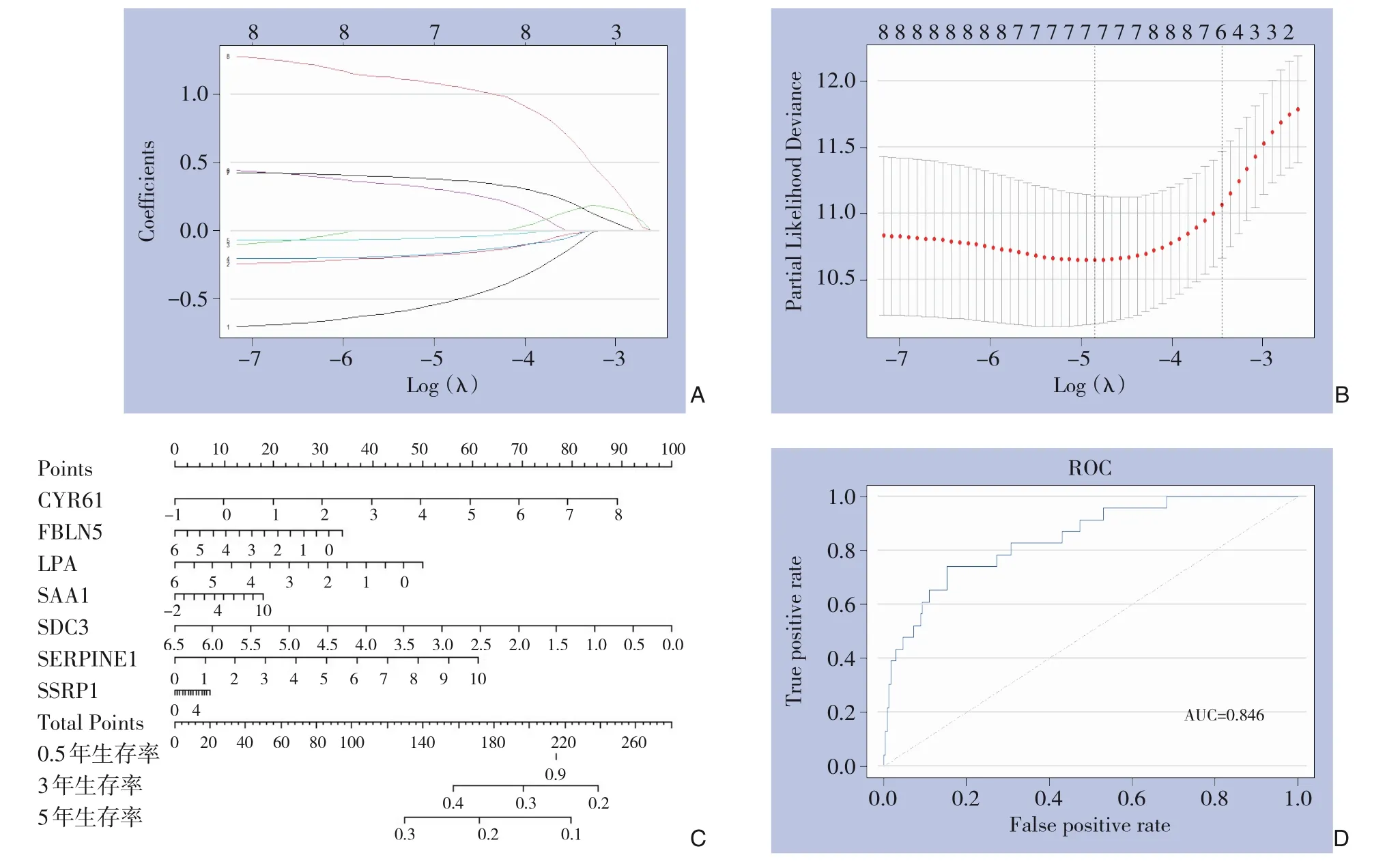
图4 构建预后模型 A:所选特征的系数由λ参数表示;B:偏似然偏差和对数(λ)用Lasso-Cox回归模型绘制;C:多因素Cox回归模型的构建及其列线图;D:ROC检测7-mRNA预后模型的预后能力Figure 4 Construction of prognosis model A: The coefficients of the selected features represented by the λ parameter; B: Partial likelihood deviation and logarithm (λ) constructed with the Lasso-Cox regression model; C: Construction of multivariate Cox regression model and its nomogram; D: Prognostic ability of the 7-mRNA signature model detected by ROC
2.4 7-mRNA模型预后能力验证分析
将风险评分高于中位数3.394 911的标记为高风险,低于中位数的为低风险,高风险样本生存时间普遍更长,截止期内死亡发生更少(图5A-B)。对比高、低风险组绘制Kaplan-Meier生存曲线,曲线显示高风险评分患者预后不良(P<0.001),高低风险评分对预后的区分度明显,与临床指标中的肿瘤大小和UICC分期具有相似的良好区分度,而比淋巴转移、远处转移和BMI值区分度要更好(图5C-H)。对风险评分、肿瘤大小和UICC分期3个指标进行Cox回归分析,风险评分无论在单因素Cox回归分析中或是在多因素Cox回归分析中都被视为独立的预后指标(P<0.001)(表2)。在考虑不同UICC分期亚型时,本研究绘制了I、Ⅱ分期和Ⅲ、Ⅳ分期两亚型的高、低风险Kaplan-Meier生存曲线,在I、Ⅱ期患者中评分对预后的区分度不理想(P>0.05),而在Ⅲ、Ⅳ期患者中有明显的区分能力(P<0.01)(图5I-J),提示本研究建立的7-mRNA预后模型更适用于较危重患者的生存结局预测。
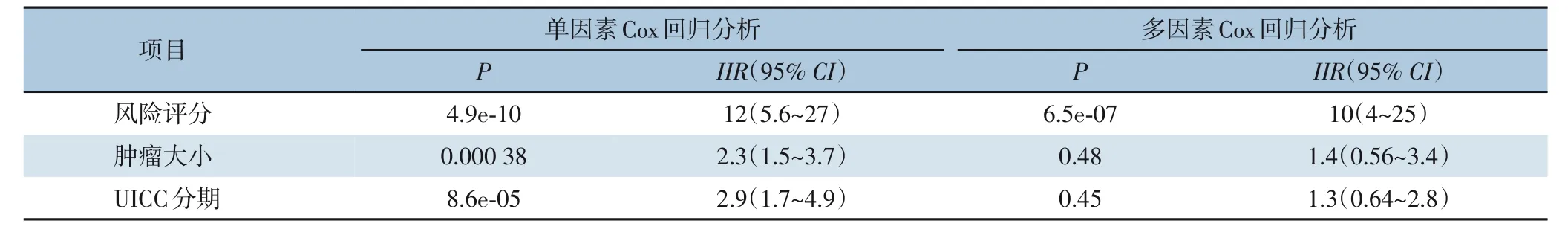
表2 与临床指标进行Cox回归分析Table 2 Cox regression analysis with clinical indicators
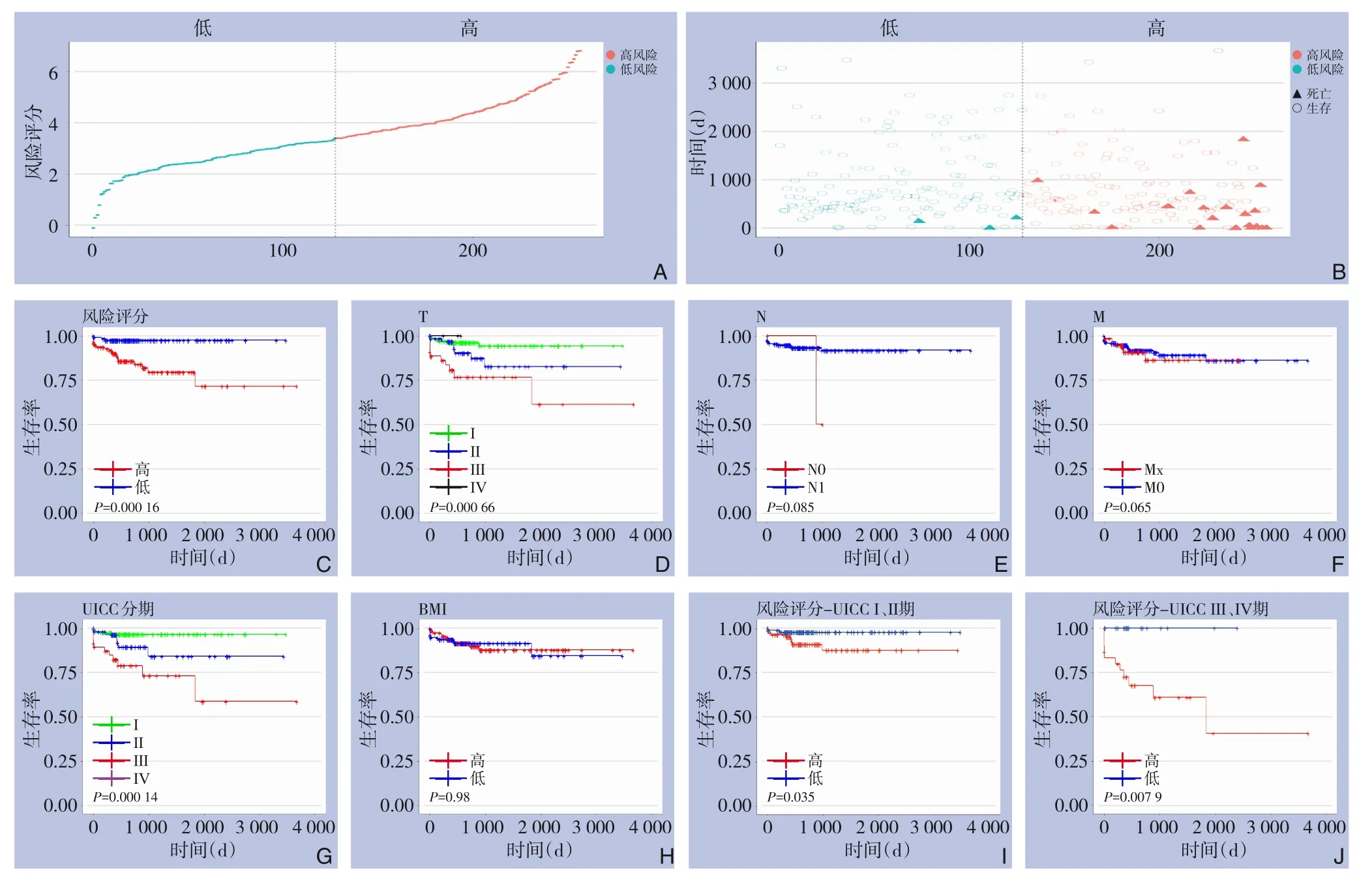
图5 7-mRNA预后模型预后能力分析验证 A-B:将风险评分高于中位数的标记为高风险,低于中位数的为低风险;C-H:按高低风险评分、肿瘤大小(T)、淋巴转移(N)、远处转移(M)、UICC分期和BMI值分别绘制的Kaplan-Meier生存曲线;I-J:两UICC亚型(I、Ⅱ期和Ⅲ、Ⅳ期)分别绘制的高低风险Kaplan-Meier生存曲线Figure 5 Analysis and verification of the prognostic ability of the 7-mRNA signature model A-B: Marking the risk score higher than the median as high risk, and the risk score lower than the median as low risk; C-H: Constructing Kaplan-Meier survival curves for the high and low-risk score, tumor size (T), lymphatic metastasis (N), distant metastasis (M),UICC stage and BMI value; I-J: Constructing high and low-risk Kaplan-Meier survival curves between the two UICC subtypes (stages I, Ⅱ, Ⅲ and Ⅳ)
2.5 分析模型的7个基因及寻找药物靶点
在424例LIHC样本中风险模型的7个基因RNA表达量在有无癌样本中差异均较明显,其中有癌样本的CYR61,FBLN5,LPA,SAA1,SDC3,SERPINE1 6个基因表达量均下调,仅SSRP1基因表达量上调(图6A)。7个基因在Cbioportal数据库样本中都存在一定程度的不同形式突变(表3)。分析风险模型的7个基因的蛋白互作信息,找到度数较高的核心基因SERPINE1和LPA(图6B)。LPA具有丝氨酸蛋白酶活性,能够自体蛋白溶解,属于纤溶酶原亚科;SERPINE1是纤溶酶原激活剂抑制剂1、丝氨酸蛋白酶抑制剂,与PLAT的快速相互作用可以作为调节纤维蛋白溶解的主要控制点。因此预测抑制纤溶酶原激活可能是HCC的新的药物靶点。
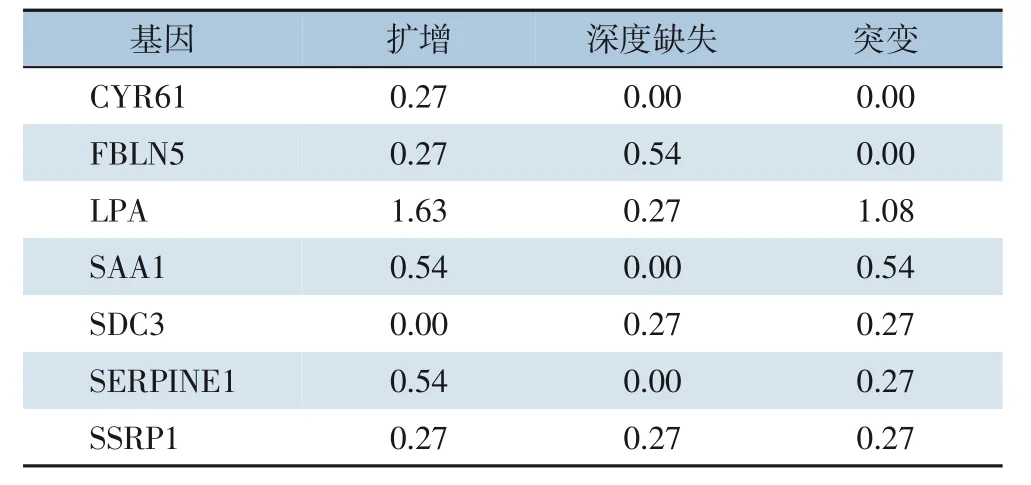
表3 模型的7个基因在372个TCGA-HCC样本中的基因变异情况(%)Table 3 Genetic variation of 7 genes in 372 TCGA-HCC samples (%)
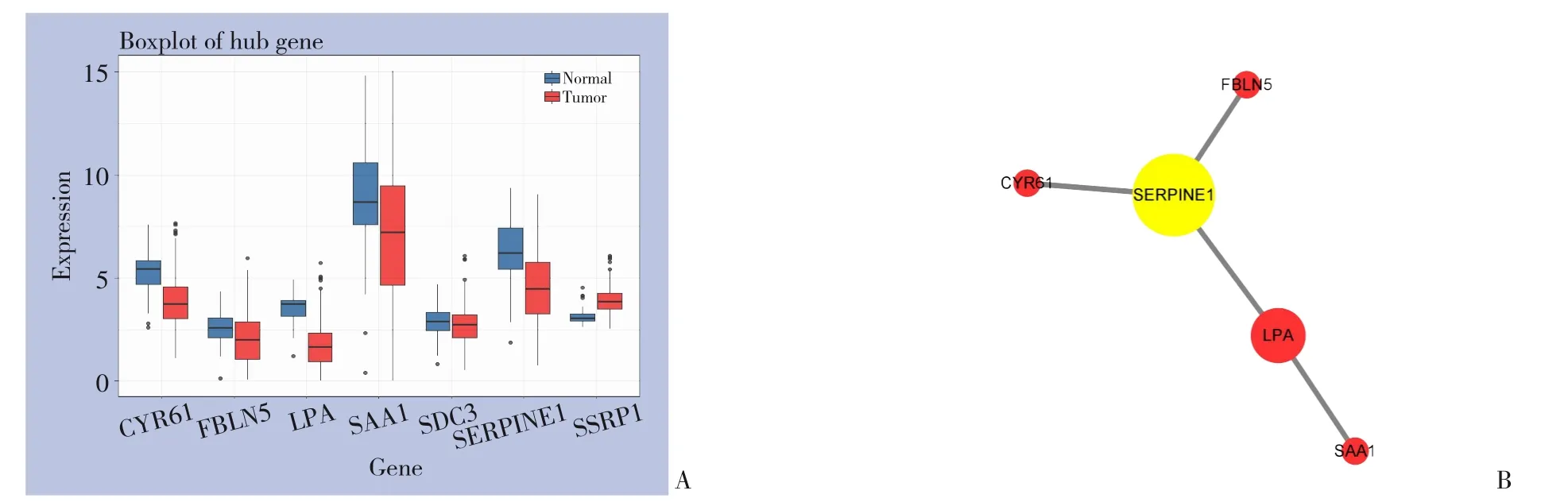
图6 模型的7个基因分析及药物靶点寻找 A:有、无癌样本7个基因表达量对比箱线图;B:蛋白互作并找出核心基因Figure 6 Analysis of seven genes and identification of drug targets A: Box-plot of comparison of 7 genes expression between cancer and non-cancer samples; B: Protein interaction and identification of the core genes
3 讨 论
在我国,HCC是最常见的肝癌类型,最近的研究表明肝硬化患者中约85%有原发性HCC发生,其5年生存率仅为18%,仅次于胰腺癌。其重要危险因素有乙型肝炎病毒(HBV)感染、过度饮酒等[3]。因此急切面临对HCC分子机制的切实解析与探究。
本研究整理了HCC表达谱数据,通过富集分析识别出了疾病相关的E2F靶点基因组和免疫相关差异基因,进一步分析得到与HCC患者预后生存相关的风险基因并构建HCC预后风险模型,并证实其具有较高的预测准确性。此外,本研究深挖了构成模型的基因,找到核心基因SERPINE1和LPA,预测了可能的治疗新靶点:纤溶酶原激活。
CDK-RB-E2F轴形成驱动细胞周期进展的核心转录机制,决定了基因组复制的时间和保真度,其最终效应子是E2F基因家族[14]。E2F靶基因包括细胞 DNA 合成及细胞周期进程的限速调解剂、原癌基因及肿瘤抑制基因等,其对许多细胞过程如细胞周期调节,血管生成,DNA损伤反应和凋亡都发挥重要作用[15],考虑到HCC是典型的多血供肿瘤,其血管生成的作用极有可能影响肿瘤自身生长与肿瘤微环境的改变,肿瘤微环境与肿瘤的发生发展、侵袭转移有密切关系。同样,作为肿瘤微环境的重要组成部分,微环境中的免疫相关细胞也对HCC的发展和进展具有重大影响,如肝动脉化疗栓塞术后调节性T细胞(Ⅲ)水平高的患者较水平低的患者的无进展生存期明显缩短[16],而主要由原发性HCC环境中预先存在的活化CD8+细胞毒性T细胞触发的PD-L1过表达则是原发性HCC的良好预后因子[17]。
目前,虽然已有较多的免疫相关生物标志物被发掘与HCC有关,但由于肿瘤微环境内基因调控与免疫浸润的复杂性,基于mRNA表达谱的生物标志物很少用于估计其在HCC中的进展。在本研究中,基因集富集分析用于识别整体和癌症发生相关性最大的基因集,而基因集单样本富集分析评分用于量化癌症样本中免疫特征的活性,并以此分数聚类以得到免疫状态不同的两个亚组,再在其中找到差异表达的基因。之后,基于上述7个基因构建了E2F靶点和免疫相关风险评分模型。值得注意的是,它能够区分高风险人群与低风险人群,并且预后估计具有较高的敏感度和特异度。在临床实用性方面,预后模型显示与患者的肿瘤大小和肿瘤UICC分期有显著相关性,且在高分期患者中的预后估计能力更为准确。因此,本研究构建的E2F靶点和免疫相关特征极有可能参与HCC的发生和进展,使其具有作为高效的临床生物标志物的潜力。尽管多基因风险评分预测能力在复杂的肿瘤微环境中有一定局限性,但具有较高预测准确性和可操作性的评分系统仍然可在筛查指南的修改、高危人群的预防中转化为临床效用[18]。
最终纳入模型的7个基因中,CYR61(富含半胱氨酸的血管生成诱导剂61)也即CCN1,属于CCN基质细胞蛋白家族,在增殖、分化、细胞凋亡、血管生成和纤维化的调节中起着关键作用[19],许多研究表明CYR61是肿瘤血管形成的关键因素,可能参与肿瘤细胞的迁移和侵袭,在多种癌症如肺癌、乳腺癌、女性生殖系统肿瘤的发生中起重要作用[20-22],但同时与本文类似的可能在非小细胞肺癌中作为肿瘤抑制因子[23];FBLN5作为母细胞糖蛋白的成员在富含弹性蛋白的组织中表达,对血管生成有抑制作用,可能通过控制细胞增殖、运动和血管生成来抑制肿瘤形成[24-25];SAA1编码血清淀粉样蛋白A,并且与C反应蛋白一起参与急性期反应,已被发现在脂质代谢中起重要作用,并有助于细菌清除,炎症调节和肿瘤发病机制[26-27];SDC3的表达在神经元、炎症性疾病,血管生成等疾病相关过程中具有重要作用[28];SSRP1沉默可影响癌症凋亡和细胞增殖[29],许多癌症相关病例中,SSRP1表达升高被证明与转移性肿瘤有关,使SSRP1成为肿瘤抑制的潜在预后标志物和抗癌靶标,与我们的结果相一致[30]。以及在蛋白互作分析中处于核心地位的SERPINE和LPA。已发表的文献表明,在胃癌中SERPINE1表达上调,增加胃癌发生风险进而影响其预后[31];在头颈部癌中SERPINE1的过表达增强肿瘤细胞迁移和侵袭,在转移发展中起关键作用[32];LPA通过促进前列腺癌中的钙网蛋白表达介导肿瘤淋巴血管生成[33],SERPINE1也曾被分别纳入自噬相关和P53途径相关的HCC预后特征与治疗靶点中[34]。因此猜测SERPINE和LPA可能通过调节血管生成和促进细胞移动影响HCC的进展。
然而,本研究仍存在一定的问题亟待解决:多基因预后模型的预测能力仍需大量多中心的循证医学证据证实;样本中截止时间内死亡样本较小可能对模型建立的准确性产生一定影响;被纳入的多基因模型的基因功能和参与的机制尚不明确,与HCC的发生、发展的关系仍需要大量的临床研究进一步印证;同时对纤溶酶原激活是如何影响HCC也需要进行更深入的研究。
利益冲突:所有作者均声明不存在利益冲突。
猜你喜欢
中老年保健(2021年3期)2021-12-03
中学生数理化·高一版(2021年2期)2021-03-19
中国生殖健康(2020年7期)2020-12-10
知识经济·中国直销(2018年8期)2018-08-23
数学学习与研究(2017年3期)2017-03-09
中国老区建设(2016年1期)2016-02-28
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年7期)2015-06-22
中国当代医药(2015年30期)2015-03-01
同位素(2014年2期)2014-04-16
