
电感耦合等离子体质谱法测定甲硝唑原料药(供注射用)中10种元素杂质含量
2023-02-14 14:01吴奔红樊华军汪秋兰武玉婧施春阳
医药导报 2023年2期
吴奔红,樊华军,汪秋兰,武玉婧,施春阳
[1.华中科技大学同济医学院附属同济医院药学部,武汉 430030;2.英格尔检测技术服务(上海)有限公司,上海 200000;3.甘肃省白银市药品检验检测中心,白银 730900]
甲硝唑为硝基咪唑类药物,对革兰阴性厌氧菌和革兰阳性菌均具有较好的抗菌活性,对原虫感染也具有较好的疗效,临床应用非常广泛,是世界卫生组织(WHO) 基本药物[1-2]。目前国内已上市的甲硝唑制剂有片剂、胶囊剂、注射剂、栓剂和凝胶剂[3]。
2020年5月国家药品监督管理局发布《关于开展化学药品注射剂仿制药质量与疗效一致性评价工作的公告》,要求对注射剂仿制药开展一致性评价,药品审评中心也发布配套指导原则[4-6],从多方面对注射剂仿制药一致性评价提出技术要求,明确要求根据国际人用药物注册技术协调会议(ICH) Q3D元素杂质指导原则的规定,对注射剂原料药的元素杂质进行评估和控制。ICH Q3D元素杂质指导原则将元素杂质分为三类,注射途径给药要求对第1类(砷、镉、汞、铅),第2(A)类(钴、镍、钒)和第3类中的锂、铜、锑10种元素进行风险控制[7]。华中科技大学同济医学院附属同济医院药学部长期从事甲硝唑原料和制剂研究[8-11],先后帮助企业获得甲硝唑原料药批准文号和欧盟CEP证书,通过甲硝唑片一致性评价,目前正在开展甲硝唑注射液一致性评价工作。笔者在本文参照USP<233>元素杂质检查法和《中华人民共和国药典》(2020年版)0412电感耦合等离子体质谱(inductively coupled plasma mass spectrometry ,ICP-MS)法指导原则,建立ICP-MS法测定甲硝唑原料药(供注射用)中10种元素,并进行了方法学验证,报道如下。
1 仪器与试药
1.1仪器 NexION 1000型电感耦合等离子体质谱仪(PE公司);WX-8000型微波消解仪(上海屹尧仪器科技发展有限公司);BSA224S型电子分析天平(德国Sartorius公司,感量:0.1 mg);UPT-II-5T型超纯水仪(Ulupure公司)。
1.2试药 标准溶液:铅(Pb)(批号:B1906159)、镉(Cd)(批号:B1912124)、钴(Co)(批号:B1902041)购于北京坛墨质检科技有限公司;汞(Hg)(批号:20A038-7)、钒(V)(批号:20A020-3)、砷(As)(批号:207003-4)、镍(Ni)(批号:208002-8)、金(Au)(批号:19C003-1)购于国家有色金属及电子材料分析测试中心;锂(Li)(批号:1094768-26)、锑(Sb)(批号:1129139-14)购于上海安谱实验科技股份有限公司;铜(Cu)(批号:R2-CU693029)购于Inorganic Ventures公司。内标:铟(117In)(批号:B1912042)购于北京坛墨质检科技有限公司;锗(74Ge)(批号:19B028)、钪(45Sc)(批号:2027033)购于国家有色金属及电子材料分析测试中心。各元素浓度均为1000 μg·mL-1。硝酸(上海凌峰化学试剂有限公司,优级纯)。自制纯化水。甲硝唑原料药(供注射用)批号为0181812001,0181912313,0181912314,由湖北省宏源药业科技股份有限公司提供。
2 方法与结果
2.1元素杂质限度的确定 参照ICH Q3D元素杂质指导原则,确定Pb、As、Cd、Hg、V、Ni、Co、Li、Cu、Sb 10种元素的注射每日允许暴露量(PDE),每日药物摄取量不超10 g的药品,限度值(μg·g-1)=PDE(μg·d-1)/药物日用量(g·d-1),见表1。
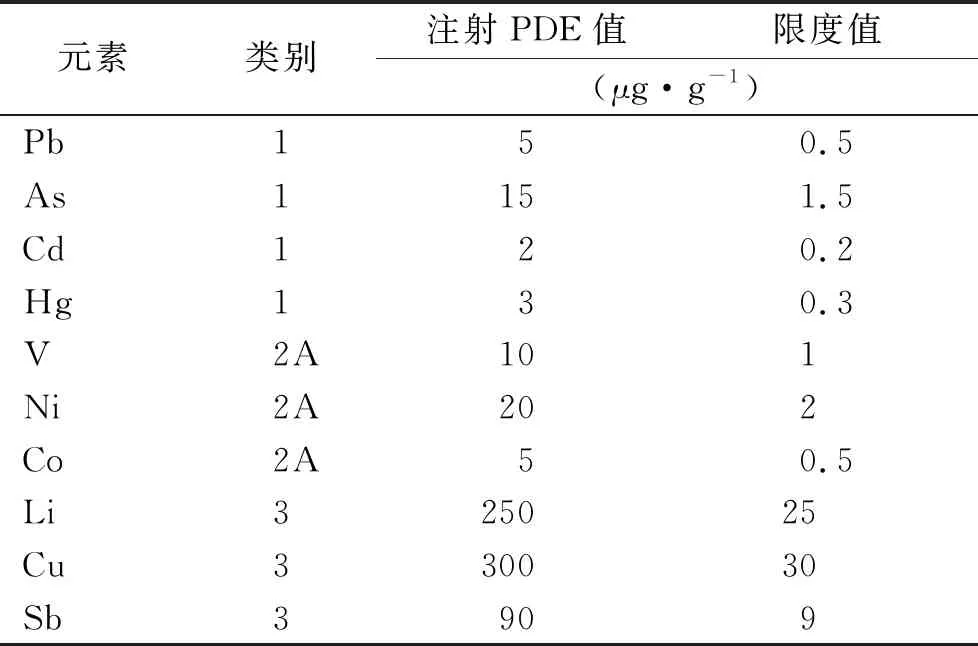
表1 元素杂质允许浓度值
2.2ICP-MS仪器条件 射频功率1 600 W,雾化器流速1.00 L·min-1,碰撞气流速4.60 mL·min-1(Pb和Hg为5.50 mL·min-1),蠕动泵转速为35 r·min-1,测量模式为碰撞池模式KED(Kinetic Energy Discrimination)。
2.3溶液制备
2.3.1标准品储备溶液制备 精密量取Pb、Cd、Hg、Co、V、Ni、As、Li、Cu、Sb标准溶液适量,分别用4%硝酸溶液定量稀释制成标准储备溶液,浓度依次为125.00,50.00,75.00,125.00,250.00,500.00,375.00,6.25×103,7.50×103,2.25×103ng·mL-1。
2.3.2内标溶液 分别精密量取74Ge、115In、45Sc标准溶液 0.2 mL,置1 L量瓶中,用4%硝酸溶液稀释至刻度,摇匀,即得混合内标溶液。
2.3.3供试品溶液和空白溶液 取本品0.25 g,精密称定,置微波消解罐中,加硝酸5 mL,100 ℃条件下预消解2 h,置微波消解仪中按120 ℃3 min,150 ℃3 min,180 ℃3 min,200 ℃30 min,依次进行消解,冷却至室温,120 ℃赶酸1 h,放冷。将消解罐内溶液转移至50 mL量瓶中,用水洗涤3次,合并洗液于量瓶中(含2.00 ng·mL-1Au),用水稀释至刻度,摇匀,即得。同法制备空白溶液。
2.3.4标准曲线溶液 分别精密量取“2.3.1”项下Pb、Cd、Hg、Co、V、Ni、As、Li、Cu、Sb标准品储备液各0.2,0.5,0.8,1.0,2.0 mL,分别置50 mL量瓶中(编号S1~S5),用4%硝酸溶液(含2.00 ng·mL-1Au)稀释至刻度,摇匀,以4%硝酸溶液(含2.00 ng·mL-1Au)为空白溶剂(编号为S0),见表2。

表2 10种成分标准曲线溶液浓度
2.4测定法 内标溶液采用在线内标加入法,样品管中依次引入标准曲线溶液、空白溶液、供试品溶液,制作标准曲线,根据标准曲线方程得到各元素杂质浓度,根据公式1计算元素杂质含量。测定Co、V、Ni、As、Cu时,以74Ge为内标元素,测定Pb、Cd、Hg、Sb时,以115In为内标元素,测定Li时,以45Sc为内标元素。含量(μg·g-1)=元素浓度(ng·mL-1)×溶液体积(mL)×10-3/称样量(g)。
2.5方法学验证
2.5.1系统适用性实验 按照“2.3.3”项下制备供试品溶液,加入各元素标准品储备溶液适量,使待测元素浓度为限度浓度,即得加标供试品溶液。在供试品溶液测定前后,分别测定加标供试品溶液 3次。结果Pb、Cd、Hg、Co、V、Ni、As、Li、Cu、Sb 10种元素的信号漂移值分别为1.31%,1.70%,6.45%,2.16%,0.70%,1.00%,2.76%,3.15%,3.19%,均<20%,表明此方法系统适用性良好。
2.5.2线性关系考察 取标准曲线溶液进样分析,以待测元素与所用内标元素响应值的比值(Y)为纵坐标,浓度(X)为横坐标,绘制标准曲线,计算回归方程。结果表明10种元素在0~200%限度范围内,线性关系良好,相关系数r均≥0.999 8。结果见表3。
2.5.3检测限与定量限 取“2.3.3”项下空白溶液进ICP-MS分析,重复测定21次,记录空白响应值(cps),计算标准偏差(δ)。根据公式(LOD=3δ/S)计算检测限(LOD),公式(LOQ=10δ/S)计算定量限(LOQ),S为各元素标准曲线斜率。结果见表3。
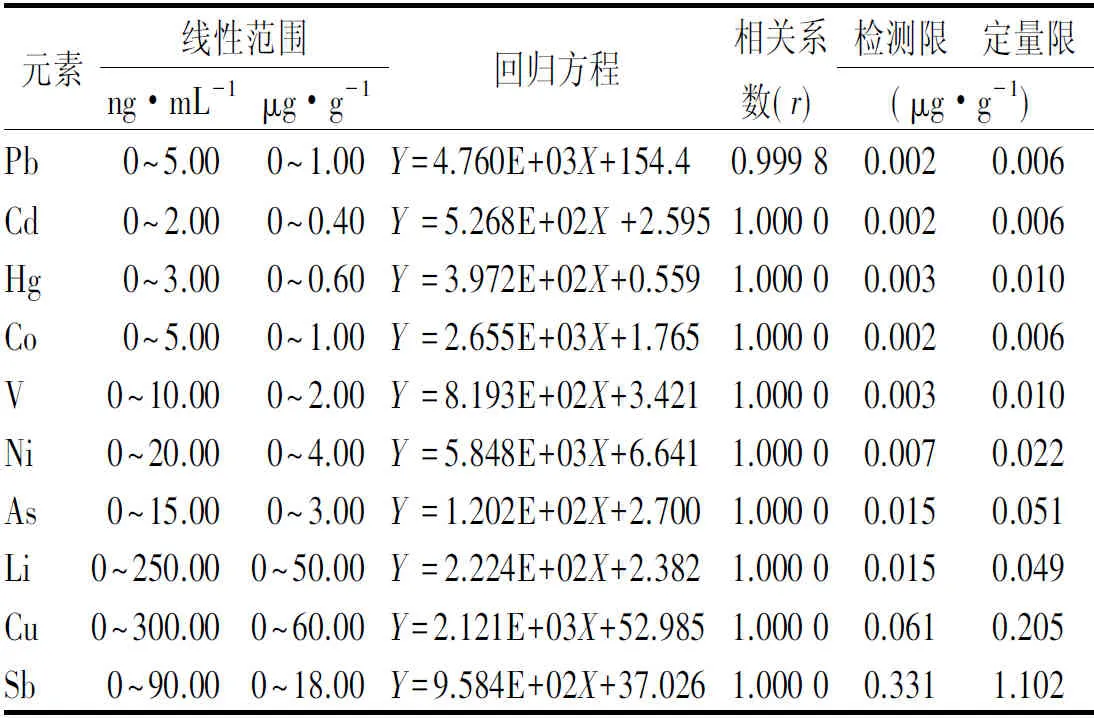
表3 10种成分回归方程、线性范围、相关系数、检测限及定量限
2.5.4重复性实验 按照“2.5.1”项下方法制备6份加标供试品溶液,进ICP-MS分析,计算各元素含量RSD(n=6)。根据USP<233>重复性要求,RSD应≤20%。结果Pb、Cd、Hg、Co、V、Ni、As、Li、Cu、Sb 10种元素的RSD分别为2.04%,5.32%,5.68%,3.39%,4.84%,2.41%,3.92%,4.25%,1.87%,4.03%,表明该方法重复性良好。
2.5.5中间精密度实验 由另一名分析人员按照“2.5.1”项下方法制备6份加标供试品溶液,进ICP-MS分析,结合重复性实验结果,计算各元素含量RSD(n=12)。根据USP<233>中间精密度要求,RSD应≤25%。结果Pb、Cd、Hg、Co、V、Ni、As、Li、Cu、Sb 10种元素的RSD分别为9.41%,5.77%,8.76%,3.59%,4.83%,3.71%,3.78%,4.99%,3.45%,9.19%,表明该方法中间精密度良好。
2.5.6准确度实验 取甲硝唑原料药(供注射用)样品(批号:0181812001)0.25 g 9份,精密称定,分别按限度浓度80%,100%,120%加入标准品储备溶液,作为低、中、高3个浓度的加标供试品溶液,每个浓度制备3个平行样品,进行加样回收实验。10种元素的回收率在76.58%~116.48%范围内,符合USP<233>中回收率在70%~150%范围内的要求。结果见表4。
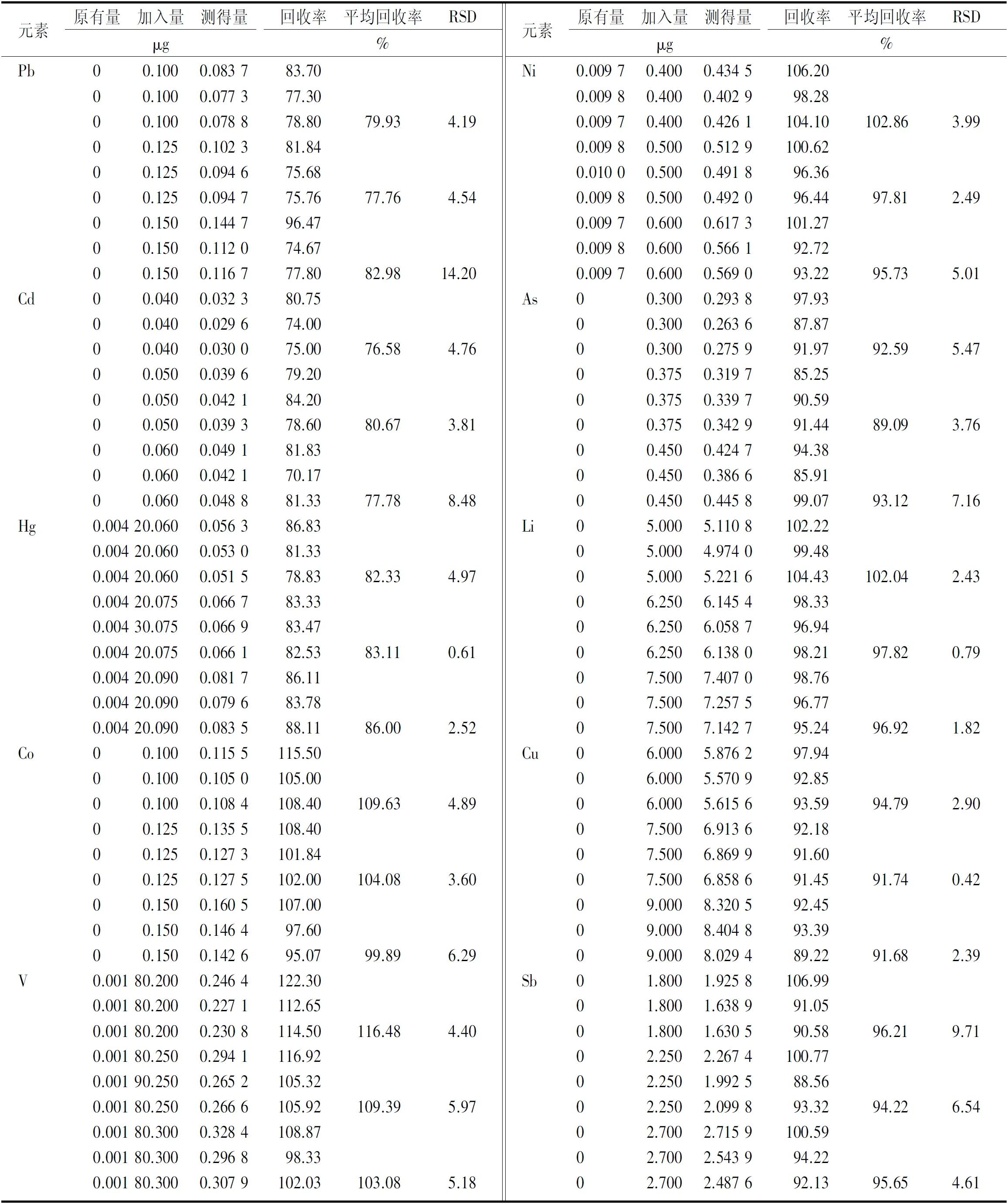
表4 10种成分回收实验结果
2.5.7耐用性实验 参照 “2.3.3”供试品溶液制备方法,将“2.3.3”项微波消解程序第4步的200 ℃下调至195 ℃,或上调至205 ℃,制备供试品溶液,再按照“2.5.1”项下制备加标供试品溶液,进行ICP-MS分析,测定各元素含量。取“2.5.1”项下加标供试品溶液,将仪器泵速由35 r·min-1下调至33 r·min-1,或上调至37 r·min-1,测定各元素含量。每个条件下测定3次,计算各元素含量RSD(n=6)。改变消解温度组中,结果Pb、Cd、Hg、Co、V、Ni、As、Li、Cu、Sb 10种元素RSD分别为1.46%,2.14%,2.55%,4.12%,5.03%,4.29%,5.27%,2.93%,1.63%,7.99%。改变泵速组中10种元素RSD分别为5.10%,5.69%,6.37%,2.87%,2.76%,2.35%,2.99%,3.63%,4.13%,5.06%,说明该方法耐用性良好。
2.5.8样品测定 取3批甲硝唑原料药(供注射用)样品,按照“2.2.3”项下方法制备供试品溶液,进行ICP-MS分析,仪器自动测试3次,取3次测量平均值,以标准曲线计算各元素含量。结果批号0181812001,0181912313,0181912314样品中Hg含量分别为0.018,0.009,0.008 μg·g-1;V含量分别为0.006,0.007,0.006 μg·g-1;Ni含量分别为0.037,0.021,0.020 μg·g-1。Pb、Cd、Co、As、Li、Cu和Sb均未检出。3批样品中各元素杂质含量远低于限度值,说明3批甲硝唑原料药(供注射用)无元素杂质风险。
3 讨论
杂质控制和分析是药品研发的重点和难点。元素杂质又称重金属,原义指比重大于5的金属,元素杂质包括可能存在于原辅料或制剂中的催化剂和环境污染物,主要在药品生产或贮藏过程中生成、加入或无意引入的物质[7]。在早期的各国药典中,对元素杂质的控制主要是针对重金属和部分无机杂质,检测方法主要包括重金属实验、炽灼残渣、硫化物和砷盐检查以及其他的化学检查法。
随着药品生产过程中催化剂和试剂应用的日益广泛,近年来元素污染的风险越来越大,研究发现某些元素杂质具有毒性,还可能对药品的稳定性、保质期带来不利影响。因此世界各国药品监管机构对药品元素杂质的控制越来越严格,各国药典经过多次更新,目前对元素杂质要求基本上和ICH Q3D保持一致。随着我国2017年加入ICH,《中华人民共和国药典》 (2020年版) 四部通则收录的9102药品杂质分析指导原则也加入元素杂质的控制要求,其规定与ICH几乎一致,提高我国药品中元素杂质控制水平和保障患者用药安全势在必行。
ICH Q3D推荐ICP-MS法和ICP-OES法为元素杂质的检测方法。ICP-OES比较,ICP-MS检测限更低,更适用于痕量和超痕量分析,谱线简单,可避免光谱干扰对测定结果的影响[12],因此本实验选择ICP-MS法。该法采用内标法校正基体效应,选择质量数和电离能与待测元素相近的元素作为内标[13],以提高分析方法的精密度。在本研究中,测定59Co、51V、60Ni、75As、63Cu时,以74Ge为内标元素,测定208Pb、111Cd、202Hg、121Sb时以115In为内标元素,测定7Li时,以45Sc为内标元素。
该法采用碰撞-反应池模式(KED)校正质谱干扰,以氦气为碰撞气体,利用干扰离子的碰撞截面比待测目标离子大,碰撞几率增加,损失更多的动能,干扰离子无法进入四级杆而达到消除干扰的目的[14]。KED消除干扰的能力受碰撞气流速的影响,不同元素需设置合适的碰撞气流速,背景等效浓度(BEC)值越低,灵敏度就越高。本研究中优化碰撞气流速时发现,当流速为4.6 mL·min-1时,Pb、Hg的BEC值较高,其他元素BEC值较低;当流速为5.5 mL·min-1时,可降低Pb、Hg的BEC值。
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02
口腔护理用品工业(2021年4期)2021-11-02
理化检验-化学分册(2020年12期)2021-01-26
云南化工(2020年11期)2021-01-14
饮食保健(2019年16期)2019-01-12
中成药(2018年6期)2018-07-11
中成药(2017年10期)2017-11-16
老年医学与保健(2017年6期)2017-02-06
现代食品(2016年24期)2016-04-28
中国粮油学报(2016年5期)2016-01-23
