
不同预处理对小鼠粪样菌群高通量测序分析结果的影响
2023-02-12 10:51:52刘昕胡会玲张敏爱薛帼珍闫晓睿张希春刘计权
生物技术进展 2023年1期
刘昕 , 胡会玲 , 张敏爱 , 薛帼珍 , 闫晓睿 , 张希春 , 刘计权
1.山西中医药大学中药与食品工程学院,山西 晋中 030600;
2.太原海关技术中心,太原 030021
肠道菌群是一个庞大的微生态系统,其所含细菌种类繁多,可分为有益菌、有害菌和中性菌3大类[1]。肠道菌群的生态平衡对于维持人体代谢、发育、免疫、感染预防及治疗等正常生理功能发挥着极其重要的作用,而这种正常的生态平衡一旦被打破,则会引起肥胖、糖尿病、肝病、免疫性疾病、精神疾病、慢性肾病等多种疾病的发生[2]。肠道菌群与人类的健康密切相关,目前已成为医学领域的研究热点。新鲜粪样经肠道排出体外,因其具有取样方便、无损伤性等特点,常被作为肠道菌群研究的取样来源[3]。
高通量测序技术因其测序快速、数据量大、能够较为全面地反应微生物群落结构等优点,已被广泛应用于肠道菌群的研究[4]。同时,DNA样品浓度、纯度和污染也是影响高通量测序结果准确度的重要因素。新鲜粪样来源于宿主肠道,除了携带宿主的肠道微生物群,同时还包含有大量的食物残渣、蛋白质、糖类、脂肪、消化酶等多种杂质,这些杂质可能会引起肠道菌群DNA发生降解、增加DNA被污染的风险、干扰PCR扩增和检测结果等问题,所以对粪样进行预处理操作,除去杂质,进而提高肠道菌群DNA的浓度和纯度,是保证高通量测序结果的有效手段[5]。已有研究报道可通过PBS缓冲液[6-7]、生理盐水[8-9]、滤器过滤[10]、均质化[11]等预处理方法达到除杂提纯的目的,其中PBS缓冲液、生理盐水预处理是常用方法,然而关于不同预处理对肠道菌群测定结果的影响少有报道。本文采用试剂盒提取DNA,通过高通量测序对原始粪样、PBS处理粪样和生理盐水处理粪样进行比较分析,以期为后续相关研究提供参考依据。
1 材料与方法
1.1 材料
1.1.1实验动物 SPF级8周龄昆明种小鼠10只,体质量22~25 g,购自北京维通利华实验动物技术有限公司。适应性饲养1周,饲养期间自由采光,给予充足的食物和水。
1.1.2试剂 胃蛋白酶购自Sigma化工有限公司;PBS缓冲液和生理盐水购自武汉普诺赛生命科技有限公司;DNA提取试剂盒和QIAamp 96 PowerFecal QIAcube HT kit(5)购自德国QIAGEN公司;Qubit DNA检测试剂盒购自Life Technologies;ExTaq高保真酶购自日本Takara公司。
1.1.3仪器 涡旋混合器购自上海第一医学仪器厂;台式高速离心机购自德国Eppendorf公司;净化工作台和立式压力蒸汽灭菌锅购自上海博讯实业有限公司;厌氧培养箱购自上海容威仪器设备有限公司;PCR仪购自美国Bio-Rad公司;电泳仪和凝胶成像仪购自Tanon公司;-80 ℃超低温冰箱购自美国Thermo Fisher Revco公司;高通量核酸纯化仪购自QIAGEN公司;高通量测序仪购自美国Illumina公司。
1.2 方法
1.2.1粪样采集及预处理 将实验小鼠分别单独放入灭菌的鼠笼中,用一次性无菌镊子,收集每个小鼠新鲜粪样2~3粒置于同一无菌离心管中,混匀。提取DNA之前,将粪样分为3组,在厌氧培养箱内迅速进行如下处理。①原始粪样组(Y):取新鲜粪样1 g,装入15 mL无菌离心管中,保存于-80 ℃冰箱。②生理盐水处理组(S):参照Garcia等[8]方法,向装有1 g新鲜粪样的50 mL离心管中加入20 mL生理盐水,用涡旋混匀仪摇动3 min后,3 000 r·min-1离心3次,每次3 min,取上清液保存于-80 ℃冰箱。③PBS处理组(P):处理液为PBS缓冲液,其余操作同生理盐水处理组。每组试验重复3次。
1.2.2样本DNA提取、PCR扩增和16S rDNA测序 DNA提取、PCR扩增和测序均由上海欧易生物医学科技有限公司完成。根据提取试剂盒所述方法提取DNA,针对V3~V4区域设计带Barcode的 特 异引物343F(5'-TACGGRAGGCAGCAG-3')和798R(5'-AGGGTATCTAATCCT-3')进行PCR扩增[12],通过Illumina MiSeq平台进行测序。
1.2.3数据分析方法 使用Trimmomatic(version 0.35)[13]软件、Flash(version 1.2.11)[14]软件、QIIME中的split-libraries(version 1.8.0)[15]软件、UCHIME(version 2.4.2)[16]软件进行微生物组学数据分析,使用QIIIME2软件进行多样性分析,使用SPSS(version 26.0)进行显著性分析,使用Metabo Analyst进行PCA分析,QIIIME2软件在门、纲、科、属水平下绘制柱状图。
2 结果与分析
2.1 测序数据的质控
2.1.1测序数据的统计 表1为3组9个样本16S rDNA V3~V4区的测序结果。由表1可知,质控之后的Clean tags数据量分布在69 909~77 752之间,Clean tags去除嵌合体后得到的Valid tags数据量分布在56 015~65 796之间,Valid tags平均长度分布在412.34~421.21 bp,各样本OTUs分布在351~670之间。

表1 3组9个样本的OTUs分析Table 1 OTUs analysis of 9 samples in 3 groups
S组3个样本有194 649条序列及1 987个OTUs;P组3个样本有192 179条序列及1 092个OTUs;Y组3个样本有186 241条序列及1 302个OTUs。
2.1.2Specaccum物种累积曲线 图1结果显示,随着样本量由小变大,曲线逐渐趋于平缓;当样本量达到8时,此时群落中的OTUs总数不再增加,表明测序深度已经基本覆盖到样品中所有的物种,测序数据量合理,测序深度足够大,可以反映绝大多数的微生物多样性信息。

图1 物种累积曲线图Fig. 1 Species accumulation plot
2.1.3Rank abundance分析 曲线越宽表明物种的组成越丰富,曲线趋势越平缓表明物种组成的均匀程度越高。图2中Rank-abundance分析结果表明,S组曲线宽度最大,说明S组小鼠粪样微生物丰度最高,而Y组和P组都有所降低;随着OTU等级增加,S组、Y组和P组曲线最终均趋于平缓,表明3组粪样菌群物种分布都较均匀,以S组物种分布的均匀度最高。

图2 等级分度曲线Fig. 2 Grade classification curve
2.1.4韦恩图 图3中心数字代表所有样本共有的OTUs,花瓣上的数字代表各个样本总OTUs减去共有OTUs的数目。由图3可知,9个样本共有188个OTUs,S组3个特异性的OTUs数目均高于P组和Y组,Y组3个特异性的OTUs数目均高于P组,表明S组的微生物多样性和丰富度最高,Y组次之,P组最低。
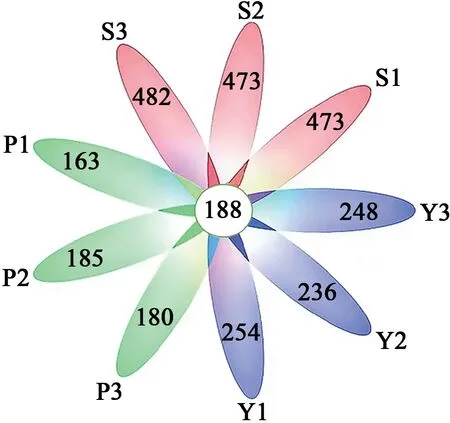
图3 Core-Pan图Fig. 3 Core-Pan diagram
2.2 不同预处理对粪样菌群多样性的影响
2.2.1AIpha菌群多样性指数分析 由表2可知,3组样本的Goods coverage均达到了0. 99以上,且3组之间无显著差异,表明本次测序的结果能真实反映样本的微生物分布情况。S组的Chao l、Observed-species、Shannon、Simpson指数均显著高于P组和Y组,Y组的Chao l、Observed-species、Shannon、Simpson多样性指数均显著高于P组,表明S组样本微生物群落的丰度和多样性最高,Y组次之,P组最低。
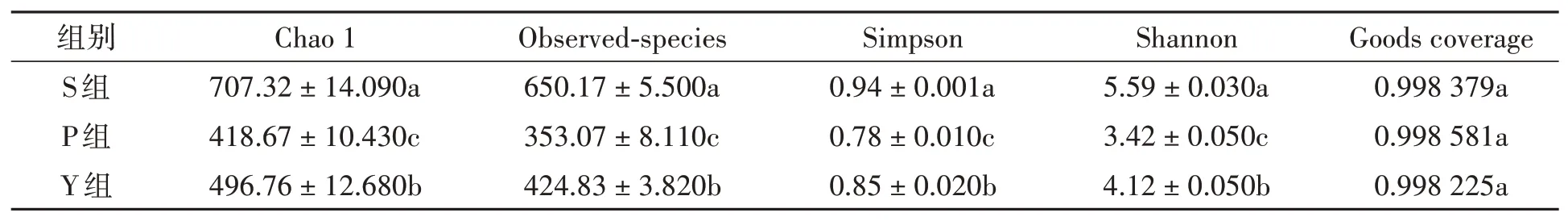
表2 不同预处理组粪样菌群的多样性指数Table 2 Diversity index of faecal flora in different pretreatment groups
2.2.2PCA分析 图4结果表明,第一、第二主坐标分别解释了菌群结构总变异的50.21%、12.27%;聚类现象表明,各组内的菌群结构相似,P组与Y组距离较近,说明P组与Y组菌群结构相近;与P组相比,Y组距离S组更近,说明Y组与S组菌群结构更相近。

图4 PCA(2D)分析Fig. 4 PCA(2D) analysis
2.2.3树状图聚类分析 采取随机重复抽样算法,对UPGMA的可靠性进行分析,构建了样本聚类树状图。由图5可以看出,3组9个样本共聚为2大类,S组3个样本单独为一类,Y组合P组的6个样本聚为一类。S组与Y组的分支距离和可靠性比S组与P组分支距离近且可靠性高;Y组部分样本与P组聚为另一类,但可靠性较低,P组与Y组部分样本中的菌群相似,但相似程度较低。
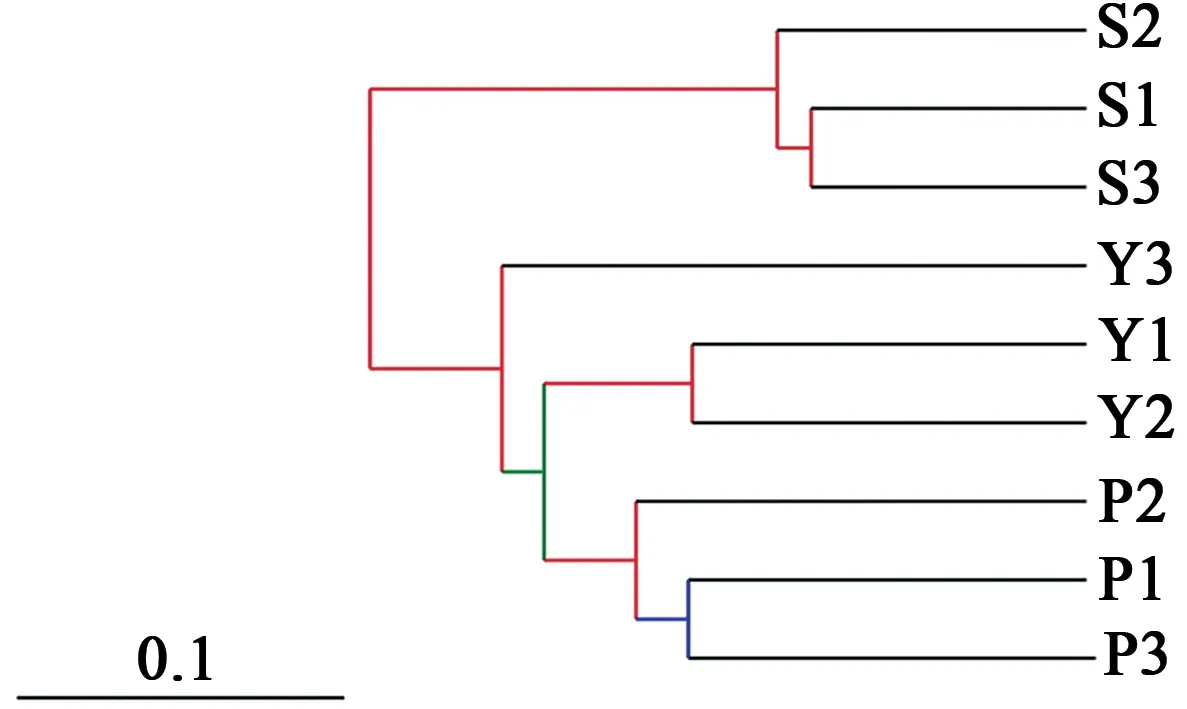
图5 Jackknifed-UPGMA样本相似性聚类图Fig.5 Jackknifed-UPGMA sample similarity cluster graph
2.3 不同预处理对粪样菌群结构的影响
2.3.1在门分类水平上的差异分析 在门水平上,S组、P组和Y组分别检出14、11、12个门,3组共有的门为11个,图6和表3列出了3组粪样丰度排在前5的共有门。拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、和变形菌门(Proteobacteria)为3个优势菌门,放线菌门(Actinobacteria)和脱铁杆菌门(Deferribacteres)含量较少。
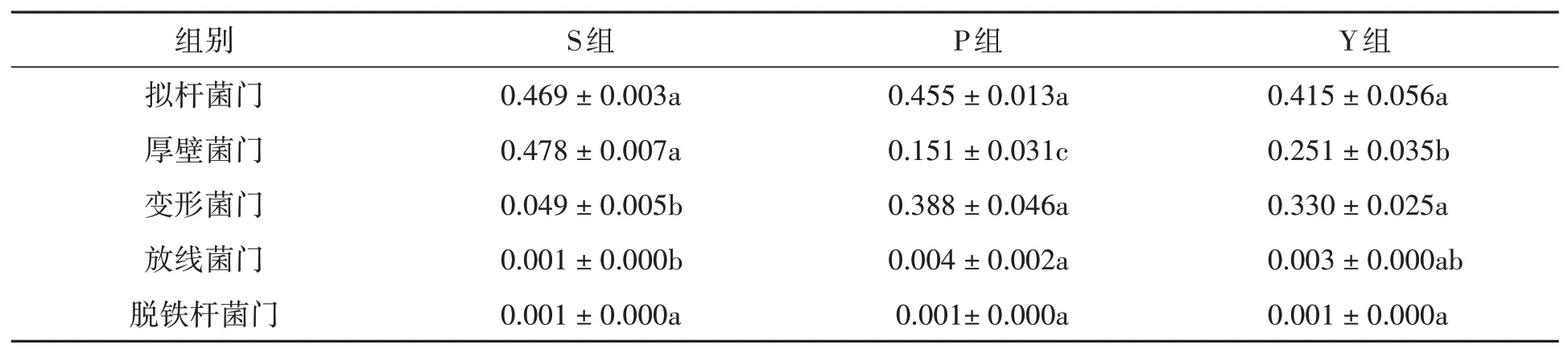
表3 在门水平上不同粪样处理组的相对丰度(Xˉ±S)Table 3 Relative abundance of fecal samples in different treatment groups at phylum level(Xˉ±S)

图6 在门水平上不同粪样处理组的群落组成分布Fig. 6 Community composition distribution in different fecal treatment groups at phylum level
拟杆菌门(Bacteroidetes)和脱铁杆菌门(Deferribacteres)的相对丰度在3组粪样之间均无显著差异(P>0.05);厚壁菌门(Firmicutes)的相对丰度在3组粪样之间差异显著(P<0.05),以S组最高,其次为Y组、P组;P组和Y组中变形菌门(Proteobacteria)的相对丰度高于S组(P<0.05),但P组、Y组之间无显著差异(P>0.05);P组中放线菌门(Actinobacteria)的相对丰度显著高于S组(P<0.05),S组与Y组之间无显著差异(P>0.05)。
2.3.2在纲分类水平上的差异分析 在纲水平上,S组、P组和Y组分别检出24、21、21个纲,3组共有的纲为20个,图7和表4列出了3组粪样丰度排在前5的共有纲,包括拟杆菌纲(Bacteroidia)、梭菌纲(Clostridia)、γ-变形菌纲(Gammaproteobacteria)、丹毒丝菌纲(Erysipelotrichia)、厚壁菌纲(Negativicutes)。

图7 在纲水平上不同粪样处理组的群落组成分布Fig. 7 Community composition distribution in different fecal treatment groups at class level
表4 在纲水平上不同粪样处理组的相对丰度(±S)Table 4 Relative abundance of fecal samples in different treatment groups at class level(Xˉ±S)

表4 在纲水平上不同粪样处理组的相对丰度(±S)Table 4 Relative abundance of fecal samples in different treatment groups at class level(Xˉ±S)
注:同一行不同组别标的不同字母表示具有统计学差异(P<0.05)。
?
拟杆菌纲(Bacteroidia)的相对丰度在3组之间无显著差异(P>0.05);S组中梭菌纲(Clostridia)、γ-变形菌纲(Gammaproteobacteria)的相对丰度显著高于P组和Y组(P<0.05),但P组和Y组之间无显著差异(P>0.05);Y组中丹毒丝菌纲(Erysipelotrichia)的相对丰度显著高于S组和P组(P<0.05),S组和P组之间无显著差异(P>0.05);厚壁菌纲(Negativicutes)的相对丰度在3组之间差异显著(P<0.05),以S组最高,其次为P组、Y组。
2.3.3在科分类水平上的差异分析 在科水平上,S组、P组和Y组分别检出62、55、59个科,3组共有26个科,图8和表5列出了3组粪样丰度排在前7的共有科,包括普雷沃氏菌科(Prevotellaceae)、肠杆菌科(Enterobacteriaceae)、毛螺菌科(Lachnospiraceae)、瘤胃球菌科(Ruminococcaceae)、伯克霍尔德科(Burkholderiaceae)、拟杆菌科(Bacteroidiaceae)、丹毒杆菌科(Erysipelotrichaece)。

表5 在科水平上不同粪样处理组的相对丰度(Xˉ±S)Table 5 Relative abundance of fecal samples in different treatment groups at family level(Xˉ±S)
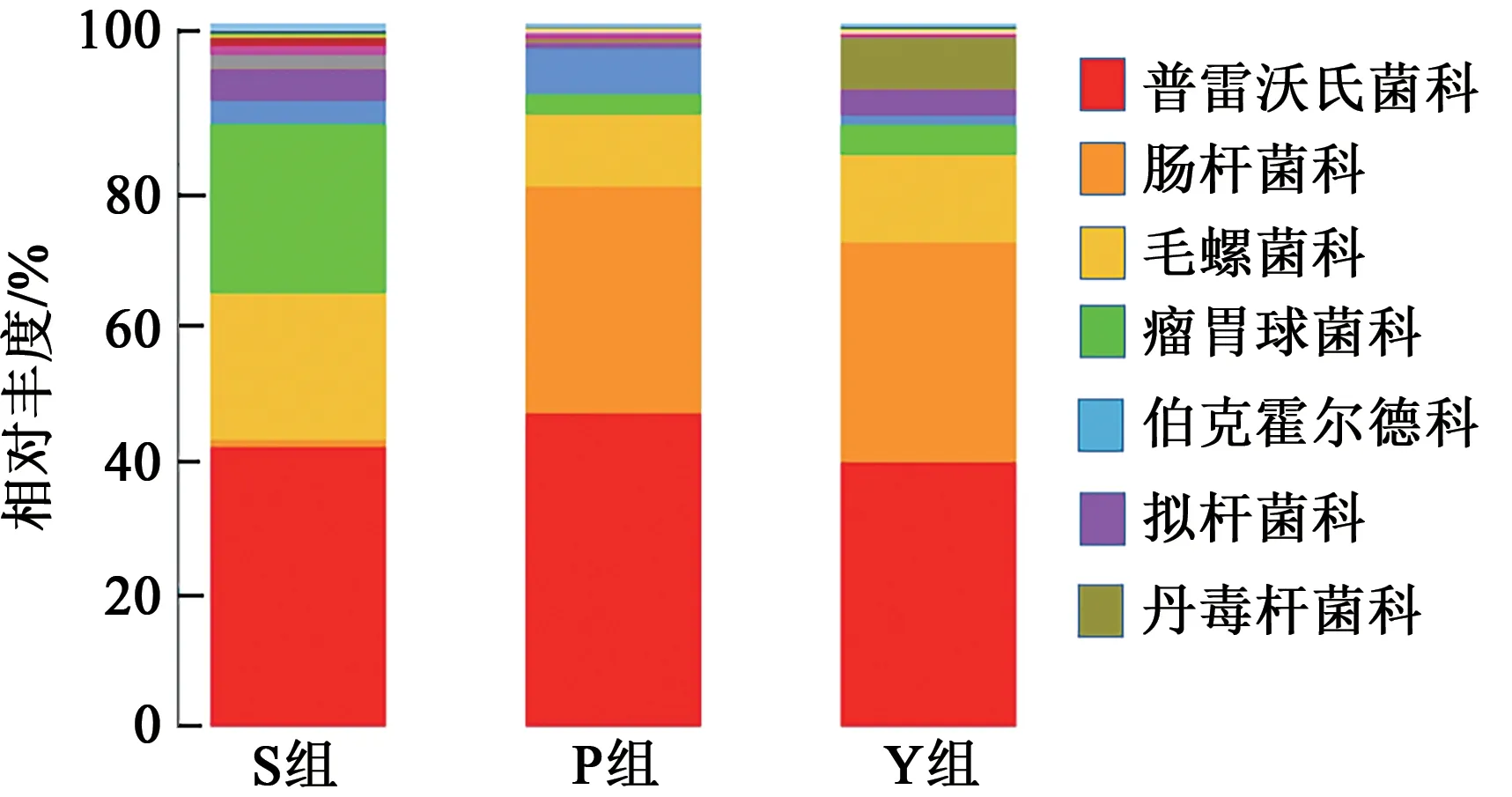
图8 在科水平上不同粪样处理组的群落组成分布Fig. 8 Community composition distribution in different fecal treatment groups at family level
普雷沃氏菌科(Prevotellaceae)的相对丰度在3组之间无显著差异(P>0.05);瘤胃菌科(Ruminococcaceae)和拟杆菌科(Bacteroidiaceae)的相对丰度在3组之间差异显著(P<0.05),以S组最高;伯克霍尔德科(Burkholderiaceae)和丹毒杆菌科(Erysipelotrichaece)的相对丰度在S组和Y之间组无显著差异(P>0.05),但均显著高于P组(P<0.05);肠杆菌科(Enterobacteriaceae)在P、Y两组的相对丰度显著高于S组(P<0.05);毛螺菌科(Lachnospiraceae)在S组的相对丰度显著高于P、Y组(P<0.05)。
2.3.4在属分类水平上的差异分析 在属水平上,S组、P组和Y组分别检出147、126、137个属,3组共有117个属,图9和表6列出了3组粪样丰度排在前10的共有属,包括普雷沃氏菌属9(Prevotella_9)、克雷伯氏杆菌属(Klebsiella)、大肠杆菌-志贺氏菌属(Escherichia_Shigella)、罗氏菌属(Roseburia)、粪杆菌属(Faecalibacterium)、副萨特氏菌属(Parasutterella)、拟杆菌属(Bacteroides)、链状杆菌属(Catenibacterium)、毛螺旋菌科UCG-004菌属(Lachnospiraceae_UCG-004)、乳梭菌属(Lachnoclostridium)。
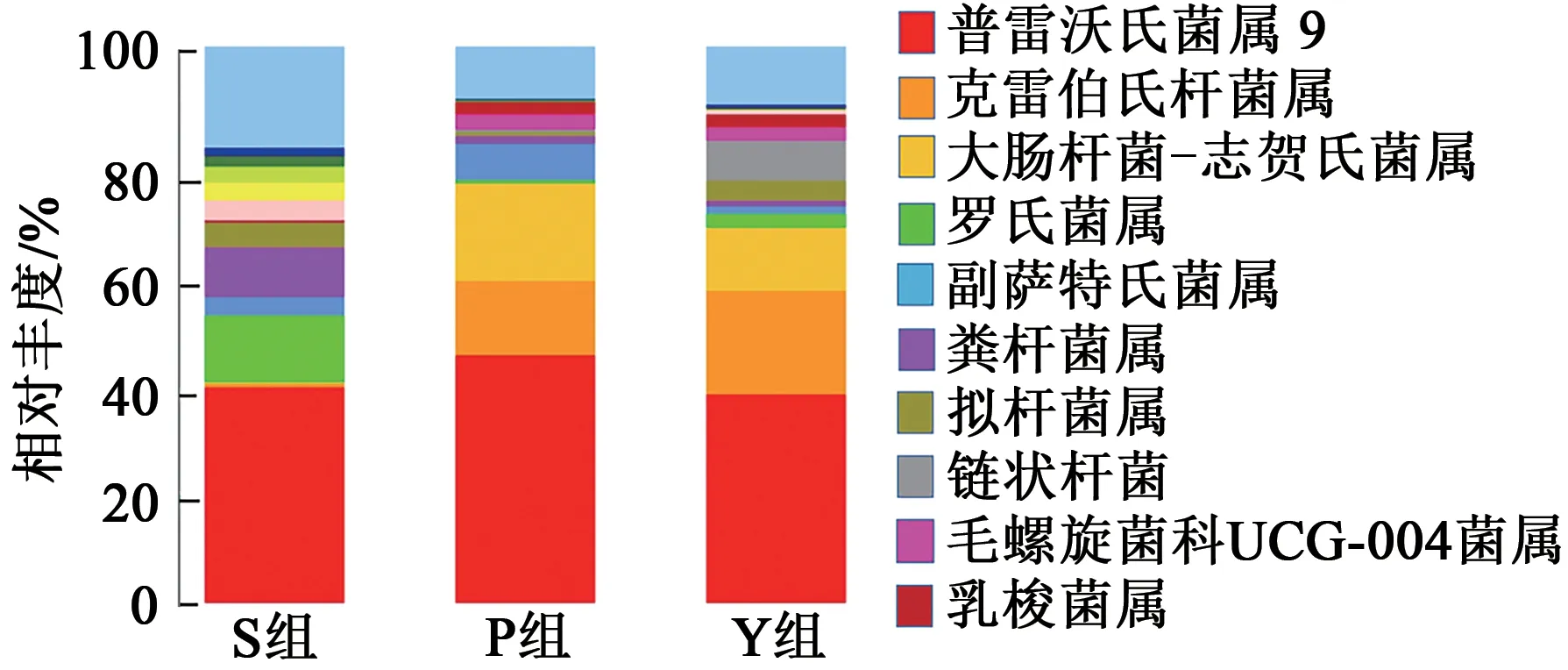
图9 在属水平上不同粪样处理组的群落组成分布Fig. 9 Community composition distribution in different fecal treatment groups at genus level
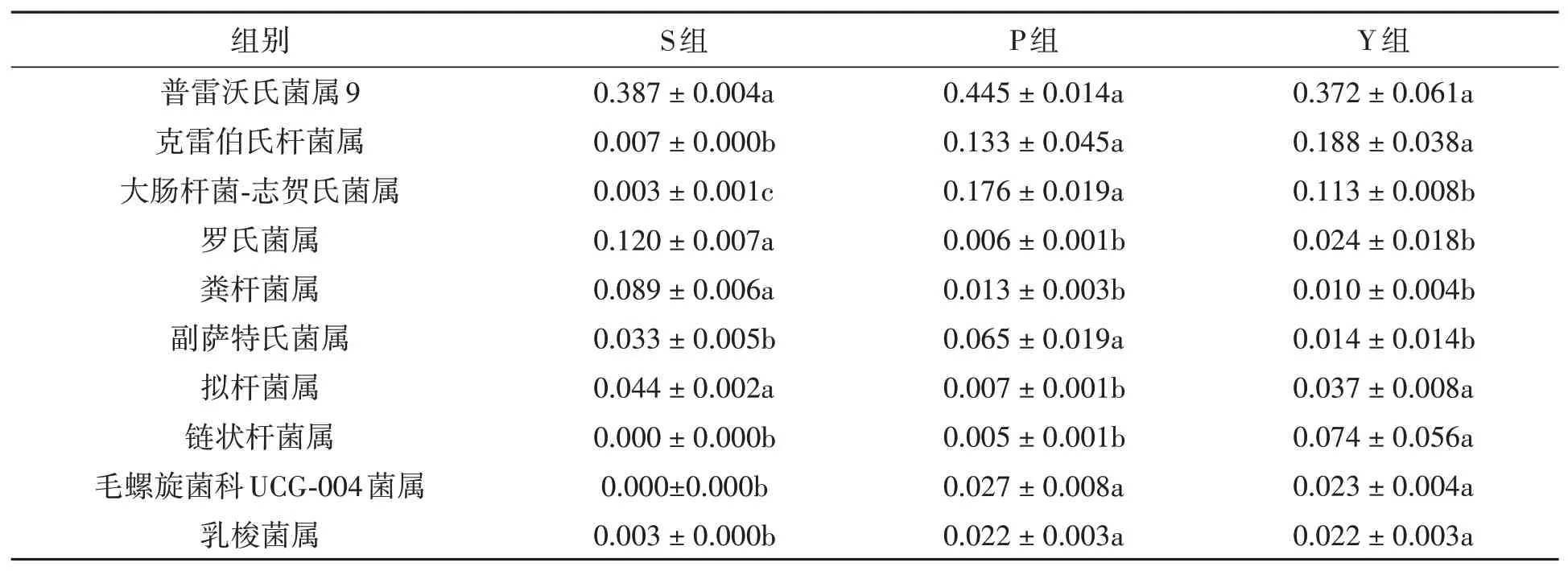
表6 在属水平上不同粪样处理组的相对丰度(Xˉ±S)Table 6 Relative abundance of fecal samples in different treatment groups at genus level(Xˉ±S)
雷沃氏菌属9(Prevotella_9)的相对丰度在3组之间无显著差异(P>0.05);P组和Y组的克雷伯氏杆菌属(Klebsiella)、毛螺旋菌科UCG-004菌属(Lachnospiraceae_UCG-004)、乳梭菌属(Lachnoclostridium)的相对丰度无显著差异(P>0.05),但均显著高于S组(P<0.05);S组罗氏菌属(Roseburia)、粪杆菌属(Faecalibacterium)的相对丰度显著高于P组和Y组(P<0.05),但P组和Y组之间差异不显著(P>0.05);大肠杆菌-志贺氏菌(Escherichia_Shigella)的相对丰度在3组之间差异显著(P<0.05),以P组最高,其次为Y组、S组。
3 讨论
高通量测序技术因其独特的优势,目前已被广泛应用于肠道菌群多样性、丰度及菌群组成的研究中。樊英等[17]利用高通量测序分析了大泷六线鱼表皮粘液及肠道内容物微生物多样性;刘铭等[18]应用高通量测序技术对我国东海带鱼肠道菌群进行了研究;申进增[19]研究了当归挥发油对自发性高血压大鼠肠道菌群的影响。肠道菌群的状态可以通过粪样菌群的变化反映出来[20],但粪样并不能真实代表肠道菌群的组成,由于受限于目前没有更好的采样方式,所以在肠道菌群的研究中,一般以新鲜粪样作为取样来源。粪样中除携带肠道细菌外,还含有大量的腐殖质、肠道内壁脱落细胞及碎片、各种有机物和无机物等[21],所以对粪样进行预处理具有必要性。董鹏飞等[9]研究发现,用生理盐水处理可以有效增加肠道菌群DNA的纯度和浓度,提高肠道微生物DNA的提取质量。张雪雁等[22]采用PBS缓冲液处理粪样,可以在一定程度上去除粪样中的杂质和抑制物。曹冉冉等[23]发现生理盐水和PBS缓冲液预处理是目前提取粪样核酸常用的方法。
本文采用Illumina Miseq高通量测序技术,对比分析原始粪样、生理盐水预处理粪样和PBS缓冲液预处理粪样中菌群的差异性,发现粪样经过生理盐水处理后,不仅可以去除杂质,同时有利于保持粪样微生物的多样性和丰富度;PBS缓冲液虽然可以去除粪样杂质,但在一定程度上降低了粪样微生物的多样性和丰度。高通量测序中,采用生理盐水对粪样进行预处理,能够在一定程度上提高粪样中菌群检测的准确性。原始粪样由于带有大量杂质可能会影响某些菌群的检出,不同溶液预处理可能会造成某些菌群的损失,不同处理方式对不同菌群的影响也不完全一样,其具体影响原因有待于进一步研究。
研究还发现,拟杆菌门、厚壁菌门和变形菌门均为原始粪样组、生理盐水处理组和PBS缓冲液处理组排在前3位的优势菌门,这与文献[24-25]报道的研究结果相一致,3组粪样均能最大程度上保留优势菌门。厚壁菌门是肠道内最为优势的一大类细菌,这一类细菌属于革兰阳性厌氧菌[19],很多厚壁菌可以产生芽孢,用以抵抗脱水和极端环境,动物肠道中的厚壁菌属于化能营养型细菌,能帮助动物从食物中获取更多的能量[26]。放线菌是革兰阳性厌氧菌,可增加肠道内抗生素的产量,进而破坏肠道菌群的稳态,加剧相关疾病的进展[27],3组粪样均检出了放线菌门。
瘤胃球菌科属于厚壁菌门,是胃肠道中分解纤维素和发酵抗性淀粉的主要菌属[28],瘤胃球菌具有碳水化合物降解活性,可通过分泌纤维素酶、半纤维素酶等降解纤维物质,并有助于细胞吸收糖分,为宿主提供所需营养物质[29];拟杆菌能够参与碳水化合物发酵、多糖代谢、胆汁酸和类胆固醇代谢、抑制机体炎性反应、影响宿主免疫系统、维持肠道正常生理等诸多功能,对宿主健康具有重要影响[30];伯克霍尔德菌科涵盖生态上极其多样化的微生物,包括真正的环境腐生生物、植物病原体、条件致病菌,多数为人类和动物的主要致病菌[31]。本次研究中,瘤胃球菌科、拟杆菌科和伯克霍尔德科作为排在前7的共有科在3组粪样均被检出。
普雷沃氏菌属能促进肠道消化碳水化合物和膳食纤维,从而有利于维持肠道菌群稳定,增强免疫功能[32];粪杆菌属、毛螺旋菌科UCG-004菌属是肠道中的有益菌属,与短链脂肪酸产生相关[33];罗氏菌属为肠道中产生丁酸的优势菌[34];拟杆菌属菌群可在人体肠道内产生乙酸、丙酸和丁酸等短链脂肪酸,而短链脂肪酸对维持机体功能具有重要作用;乳梭菌属在肠道中多数为非致病菌,少数为致病菌;此外,克雷伯氏菌、大肠杆菌-志贺氏菌均来自肠杆菌科,属于条件致病菌[35]。以上菌属作为3组粪样丰度排在前10的共有属均被检出。
肠道菌群是一个极其复杂的微生态系统,包括许多生物因子和非生物因子,不同的取样方法、预处理方式、DNA提取方式、测序方式等均会对肠道菌群的研究结果产生影响,同一处理方式对不同肠道微生物的的影响也可能不同。此外,小鼠物种、个体、周龄等差异均会对肠道菌群研究结果产生影响,今后应对影响肠道菌群结构的可能因素进行系统分析。
猜你喜欢
野生动物学报(2023年2期)2023-05-16 03:06:28
国际太空(2023年1期)2023-02-27 09:03:42
疯狂英语·初中天地(2022年5期)2022-07-06 02:29:38
疯狂英语·初中版(2022年5期)2022-05-11 03:08:06
野生动物学报(2021年2期)2021-05-06 03:10:14
透析与人工器官(2020年1期)2020-11-16 01:42:34
铁道通信信号(2019年8期)2019-10-10 05:06:00
中学生数理化·八年级物理人教版(2018年5期)2018-06-21 08:01:42
养猪(2018年3期)2018-06-12 07:01:22
中国发展观察(2017年8期)2017-04-26 03:51:50
