
贵州传统小曲酵母菌分子指纹图谱分析
2023-02-07 06:48王春晓何宇淋唐佳代邱树毅
食品科学 2023年2期
王春晓,何宇淋,唐佳代,2,邱树毅
(1.贵州大学酿酒与食品工程学院,贵州省发酵工程与生物制药重点实验室,贵州 贵阳 550025;2.茅台学院酿酒工程系,贵州 仁怀 564500)
小曲是中国传统四大发酵制剂之一,其以米粉为原料且体积小,主要用于酿造米酒、黄酒和白酒(米香型、清香型和豉香型)[1]。贵州米酒和小曲白酒酿造中至今仍多采用传统小曲,形成了酒的独特品质风味,如贵州少数民族的九仟酒、黑糯米酒、咂酒等,但贵州传统小曲中功能微生物的研究报道较少,尤其需关注主要酒化和酯化等作用的功能酵母菌的研究和开发[1]。近年来,随着高通量测序技术的兴起,小曲中主要酿酒微生物的多样性研究得以快速推进[2-9],然而高通量测序在种水平的分类鉴定依赖于已建立的基因数据库如GenBank等[10],基因数据库中缺乏的菌种序列信息将会导致高通量测序结果中无法分类的微生物群体存在,本课题组在贵州传统小曲的高通量测序分析中发现在菌种水平上无法分类的真菌微生物群体最高可达近50%,因受测序引物影响其中无法分类的酵母真菌比例无法确知[3]。总之,依赖于培养的功能酵母菌种水平序列分析有助于基因数据库的完善,而分子指纹图谱分析则为功能酵母菌的开发利用提供可追溯的分子身份。本研究在传统分离培养和26S rRNA基因D1/D2区域序列分析的基础上,进一步采用5.8S rRNA基因ITS区限制性片段长度多态性分析(restriction fragment length polymorphism analysis of the ITS region of 5.8S rRNA gene,5.8S-ITS-RFLP)和串联重复-tRNA(tandem repeattRNA,TRtRNA)指纹图谱法对贵州传统小曲中的酵母菌进行菌种水平和种内分子指纹图谱分析,旨在为功能酵母菌的开发利用提供分子理论基础。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株
59 株酵母菌分离自贵州不同地区2017年生产的传统小曲,其中4 株分离于贵州省西北部威宁县,菌株编号为FBKL2.8048、FBKL2.8049、FBKL2.8055和FBKL2.8056;11株分离于贵州省西南部盘州市,菌株编号为FBKL2.8027、FBKL2.8029~FBKL2.8032、FBKL2.8034、FBKL2.8050~FBKL2.8054;18株分离于贵州省西南部兴仁市李官,菌株编号为FBKL2.8001~FBKL2.8009、FBKL2.8011、FBKL2.8012、FBKL2.8022~FBKL2.8024、FBKL2.8041、FBKL2.8042、FBKL2.8057、FBKL2.8058;14株分离于贵州省西南部兴仁市肖家湾,菌株编号为FBKL2.8015、FBKL2.8017~FBKL2.8019、FBKL2.8021、FBKL2.8028、FBKL2.8059~FBKL2.8066;12株分离于贵州省北部遵义市,菌株编号为FBKL2.8026、FBKL2.8035~FBKL2.8040、FBKL2.8043~FBKL2.8047[3]。所有菌株均由贵州大学酿酒与食品工程学院贵州省发酵工程与生物制药重点实验室保存。
1.1.2 培养基
酵母浸出粉胨葡萄糖(yeast extract peptone dextrose,YPD)培养基:1%酵母浸粉,2%葡萄糖,2%蛋白胨,2%琼脂粉[11]。将酵母菌划线培养于YPD培养基上,30 ℃培养2 d后用于提取酵母菌DNA。
WL营养琼脂培养基:根据上海博微生物科技有限公司提供的说明配制[12-13],将酵母菌划线培养于WL培养基上,30 ℃培养5 d后,用于观察记录形状、色泽、边缘和质地等菌落特征。
1.1.3 试剂
引物、限制性内切酶、TaqDNA聚合酶、DNA Marker 宝日医(TaKaRa)生物技术有限公司;HyAgarose琼脂糖 厦门太阳马生物工程有限公司;核酸染料Genegreen 天根生物科技(北京)有限公司。
1.2 仪器与设备
LS-B75L-I立式压力蒸汽灭菌器 江阴宾江医疗设备有限公司;SW-CJ-ID单人超净工作台 苏州净化设备有限公司;SPX-250-Z生化培养箱 上海博迅实业有限公司医疗设备厂;Allegra X-30R离心机 美国Beckman Coulter公司;MIT-100恒温混匀仪 杭州米欧仪器有限公司;IMS-20制冰机 常熟市雪科电器有限公司;JXFSTPRP-24细胞破碎仪 上海净信发展有限公司;PHS-3C精密酸度计 上海大普仪器有限公司;Bio-Bset140E凝胶成像仪 美国SIM西蒙国际公司;CFX Connect Real-Time System聚合酶链式反应(polymerase chain reaction,PCR)仪、核酸电泳仪美国伯乐仪器设备公司。
1.3 方法
1.3.1 酵母菌种水平鉴定
1.3.1.1 酵母菌DNA提取
取适量酵母菌体采用石英砂破壁法提取DNA[14-15]。
1.3.1.2 26S rRNA 基因D1/D2区域序列分析
26 SrRNA 基因D1/D2 区测序分析的PCR体系参照Wang Chun xiao 等[16]方法,采用NL1(5’-GCATATCAATAAGCGGAGGAAAAG-3’)和NL4(5’-GGTCCGTGTTTCAAGACGGG-3’)作为引物,PCR扩增程序参考田进等[17]。PCR扩增产物经1.5%琼脂糖凝胶电泳进行检验,电泳液为0.5×TBE缓冲液,电泳条件为90 V、50 min,使用凝胶成像仪查看电泳结果并参考100 bp DNA Marker读取条带大小,经检验大小为600 bp左右的单一PCR扩增产物送至北京六合华大基因科技有限公司进行纯化并以NL1为引物开展测序,去除序列前段的杂峰序列后,采用BLAST(https://blast.ncbi.nlm.nih.gov/Blast.cgi)与GenBank数据库中的模式菌株序列进行比对,以相似性最高的菌种作为菌种水平鉴定结果[3]。本研究进一步使用MEGA5.0构建所有测序菌株和模式菌株的系统发育树,具体参数为采用引导程序法进行系统发育实验,引导程序重复数量为1000,置换模型为Kimula双参数法[17]。
1.3.1.3 5.8S-ITS-RFLP分析
5.8S-ITS-RFLP 分析的PCR 体系参照Wang Chunxiao 等[16]方法,采用ITS1(5’-TCCGTAGGTGAACCTGCCG-3’)和ITS4(5’-TCCTCCGCTTATTGATATGC-3’)作为引物,PCR扩增程序参考田进等[17]方法。PCR扩增产物电泳条件和谱带读取如1.3.1.2节所述,并进一步采用限制性核酸内切酶(HaeIII、MboII、DdeI和HinfI)对PCR扩增产物进行酶切,酶切反应体系参考田进等[17]方法,酶切产物检测如1.3.1.2节所述。将读取图谱条带按“0”(无)和“1”(有)进行标注,使用Origin 2019软件进行系统聚类分析,分析方法为多变量分析,聚类方法为平均法,距离类型采用欧式距离。
1.3.2 TRtRNA指纹图谱分析
TRtRNA指纹图谱分析的PCR体系和扩增程序参考田进等[17]方法,采用两对引物开展PCR扩增,引物对一为5CAG(5’-CAGCAGCAGCAGCAG-3’)和TtRNASc(5’-GCTTCTATGGCCAAGTTG-3’),引物对二为ISSR-MB(5’-CTCACAACAAC AACAACA-3’)和TtRNASc[18]。PCR扩增产物电泳条件如1.3.1.2节所述,谱带读取参照100 bp和250 bp DNA Marker。如1.3.1.3节所述开展谱带的系统聚类分析。
2 结果与分析
2.1 酵母菌种水平分析
贵州不同地区传统小曲中分离的59 株酵母菌在WL培养基上呈8 类菌落特征(图1),其中I类和II类菌落都易挑起,I类呈奶油圆锥形白色凸起,II类表面不光滑、呈蜡质蓝绿色凸起,III~VIII类的共同特征为都不易挑起,表面呈现不同特征的短绒毛凸起,III类呈白色而IV类至VIII类具浅绿或蓝绿边缘。

图1 贵州传统小曲酵母菌落特征图Fig.1 Colony characteristics of yeasts isolated from traditional Guizhou Xiaoqu
59 株酵母菌经26S rRNA基因D1/D2区域序列分析,通过与模式菌株的同源序列比对确定菌种水平分类地位[3]:1 株T.asahii、32 株S.fibuligera、8 株S.malanga、4 株H.burtonii、6 株W.anomalus和8 株S.cerevisiae。其系统发育分析结果如图2所示,其中T.asahii与其余5 种酵母菌的遗传关系最远,S.fibuligera和S.malanga作为同属内不同菌种展现了较近的遗传关系(序列比对相似性为93.12%),而二者与H.burtonii、W.anomalus和S.cerevisiae之间的遗传关系相对较远。此外,4 株H.burtonii与模式菌株之间体现了一定的遗传距离(菌株FKBL2.8062与模式菌株之间序列比对相似性为99.6%,而其余3 株菌与模式菌株之间的序列比对相似性为99.8%,图2)。

图2 贵州传统小曲分离酵母菌与模式菌株基于26S rRNA基因D1/D2区域序列构建的系统发育树Fig.2 Phylogenetic tree of yeasts isolated from traditional Guizhou Xiaoqu and type strains based on the D1/D2 domain of the 26S rRNA gene sequence
为贵州传统小曲酵母菌种水平鉴定提供简单快捷的方法,本研究进一步采用5.8S-ITS-RFLP分析方法对59 株酵母菌的ITS区进行PCR扩增和酶切(表1),PCR扩增产物大小展示了6 个菌种酵母菌之间的明显与微弱差异(图3),如6 株W.anomalus和32 株S.fibuligera的扩增产物大小都为600 bp,因此进一步将PCR扩增产物开展4 种限制性内切酶酶切分析(表1,图4),PCR扩增产物结合HaeIII和HinfI酶切可清晰将S.cerevisiae、S.malanga与H.burtonii从6 个菌种中区分出来,而W.anomalus、S.fibuligera与T.asahii菌种的5.8S-ITS-RFLP分析最好采用PCR扩增产物结合MboII和DdeI酶切。

表1 贵州传统小曲中分离酵母的5.8S-ITS-RFLP分析结果Table 1 Results of 5.8S-ITS-RFLP analysis of yeasts isolated from traditional Guizhou Xiaoqu

图3 6 种酵母ITS区扩增图谱Fig.3 Electrophoretogram of ITS region amplified from six yeast species
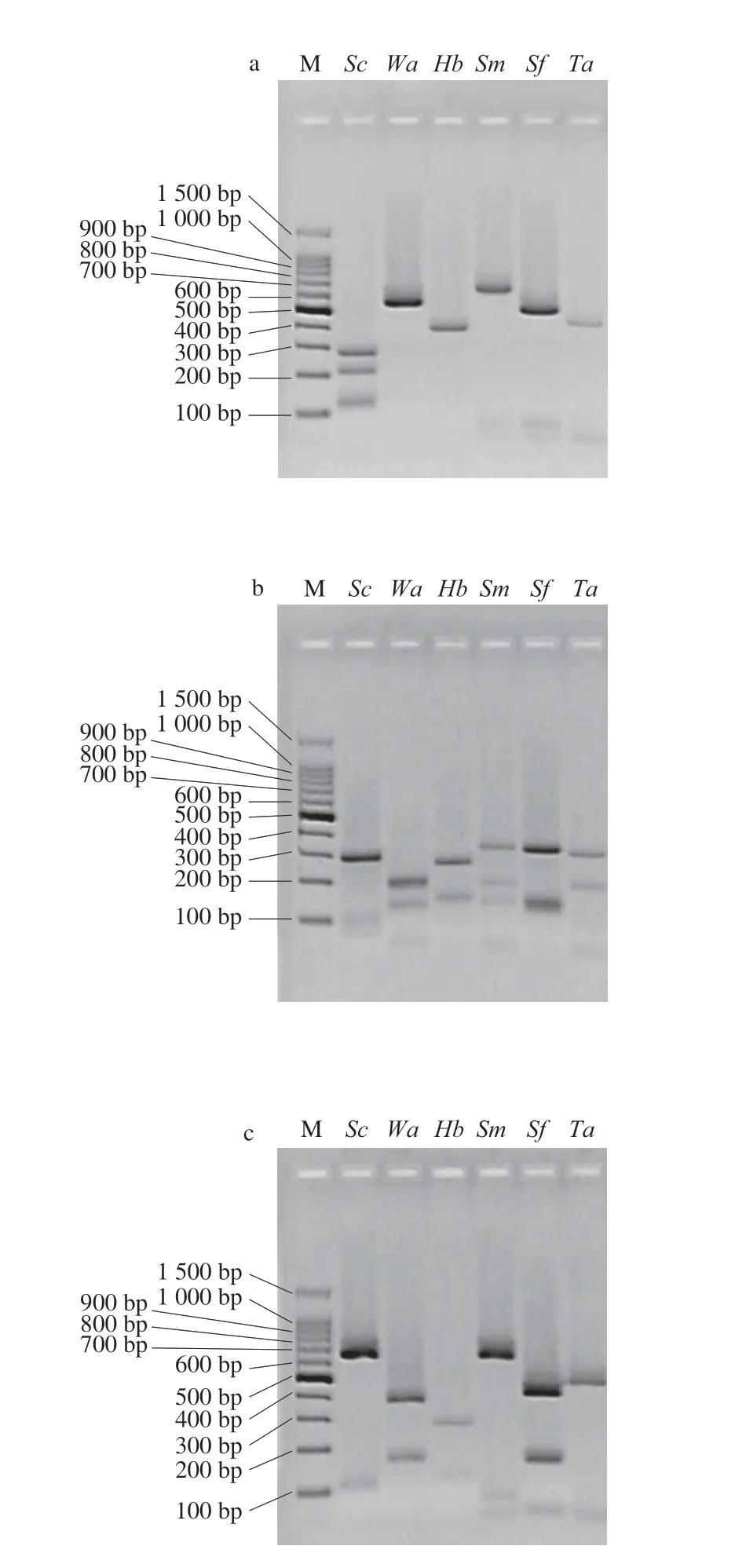

图4 6 种酵母5.8S-ITS-RFLP酶切图谱Fig.4 Electrophoretograms of six yeast species with restriction enzyme digestion and 5.8S-ITS-RFLP analysis
本研究将5.8S-ITS-RFLP分析方法中6 个菌种的全部限制性内切酶(HaeIII、MboII、HinfI和DdeI)酶切图谱结果进行聚类分析(图5),遗传距离为2.1左右可以将6 个菌种区分开来,且S.malanga和S.fibuligera菌种之间的遗传距离近于其他4 个酵母菌种,而S.cerevisiae与T.asahii与其他4 个酵母菌种之间的遗传距离较远。

图5 6 种酵母菌株5.8S-ITS-RFLP酶切结果聚类分析树状图Fig.5 Dendrogram of cluster analysis of 5.8S-ITS-RFLP profiles of six yeast species
2.2 TRtRNA指纹图谱分析
采用两对微卫星引物对59 株酵母菌株进行扩增图谱差异分析。引物对TtRNASC和ISSR-MB的扩增图谱将全部酵母菌株分为11 类(图6,下滑线标识亮条带):a类图谱条带为410 bp,包含8 株S.cerevisiae;b1类为480 bp,包含3 株H.burtonii;b2类图谱条带为500 bp,有1 株H.burtonii;c1类为4500、1500、1300、1000、620、110 bp,有4 株W.anomalus;c2类为4500、1500、1200、1000、620、500、350、200、110 bp,包含2 株W.anomalus;d类为2700、2000、1000、500、450、110 bp,包含8 株S.malanga;e1和e2类分别为2300、1500、1400、1000、900、420、350、200、160 bp和1000、900、420、350、200、160 bp,分别有1 株和2 株S.fibuligera;e3类为1000、900、420、350、200 bp,有14 株S.fibuligera;e4类为2300、1400、950、800、420、400、350、250、200、160 bp,包含10 株S.fibuligera;e5类为1000、900、500、420、350、200 bp,包含5 株S.fibuligera;T.asahii无扩增图谱因此未计入类别。11 类图谱条带的聚类分析显示e1、e2、e3和e5类遗传关系较近,a、b1和b2类遗传关系较近,两者内部遗传距离小于2.1,而e4类与其余所有类别遗传距离最远(遗传距离>3,图6)。
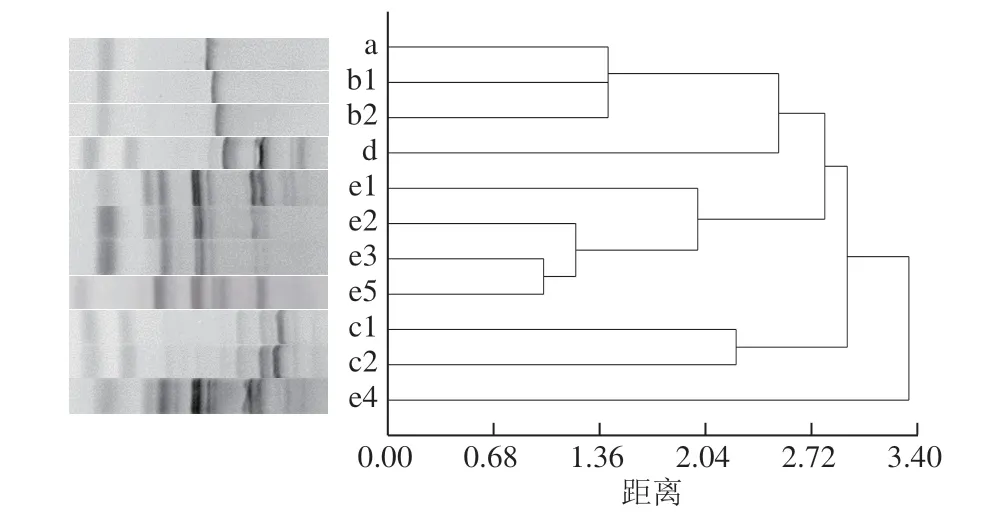
图6 引物对TtRNASC和ISSR-MB的图谱分析结果Fig.6 Analysis of electrophoresis profiles using primer pairs of TtRNASC and ISSR-MB
引物对TtRNASC和5CAG将全部酵母菌株分为11 类(图7,下滑线标识亮条带):A类图谱条带为2000、1300、1000、700、500、160、100 bp,包含8 株S.cerevisiae;B类为1300、1100、1000、500、450、300、110 bp,有1 株H.burtonii;C1、C2和C3类分别为2100、1000、900、700、600、300、280、120 bp,3000、2000、1100、1000、700、600、400、300、110 bp和3000、2000、1300、1100、1000、400、300、100 bp,分别包含1 株H.burtonii;D1类为3000、1400、1200、1000、750、700、310、290 bp,包含5 株W.anomalus;D2类为1200、800、700、660 bp,有1 株W.anomalus;E类为900、750、660、480、450、400、310、280、200、150、100 bp,有8 株S.malanga;F1类为1100、1000、680、600、580、400、190 bp,包含16 株S.fibuligera;F2类为1200、800、700、680、580、400、210、200 bp,包含9 株S.fibuligera;F3图谱条带为680、600、580、400、190 bp,包含7 株S.fibuligera;T.asahii为250 bp未计入类别。11 类图谱条带的聚类分析显示,E类明显区别于其他菌株类型,遗传距离接近4,F1类和F3类遗传关系最近(图7)。
将TRtRNA指纹图谱分析采用两对引物获取的全部图谱类型进行结合分析,可将58 株酵母菌分为17 个基因型(图8):8 株S.cerevisiae被鉴定为基因型1(图谱型为a-A),4 株H.burtonii被鉴定为基因型2(图谱型为b1-B,菌株编号FBKL2.8064)、3(图谱型为b1-C1,菌株编号FBKL2.8018)、4(图谱型为b1-C2,菌株编号FBKL2.8021)、5(图谱型为b2-C3,菌株编号FBKL2.8062),6 株W.anomalus被鉴定为基因型6(图谱型为c1-D1,4 株)、7(图谱型为c2-D1,菌株编号FBKL2.8023)、8(图谱型为c2-D2,菌株编号FBKL2.8024),8 株S.malanga被鉴定为基因型9(图谱型为d-E),32 株S.fibuligera被鉴定为基因型10(图谱型为e1-F1,菌株编号FBKL2.8026)、11(图谱型为e2-F3,菌株编号FBKL2.8041)、12(图谱型为e3-F1,7 株分离于盘州市小曲,6 株分离于遵义市小曲,1 株分离于威宁县小曲)、13(图谱型为e4-F1,菌株编号FBKL2.8048)、14(图谱型为e4-F2,4 株分离于盘州市小曲,2 株分离于李官小曲,3 株分离于威宁县小曲,1 株分离于肖家湾小曲)、15(图谱型为e2-F1,菌株编号FBKL2.8035)、16(图谱型为e5-F1,菌株编号FBKL2.8036)、17(图谱型为e5-F3,3 株分离于遵义市小曲,1 株分离于李官小曲)。1 株T.asahii因缺失引物对TtRNASC和ISSR-MB扩增结果,因此未计入基因型分析。17 个基因型的聚类分析结果(图8)表明,遗传距离大于3.6以上时TRtRNA指纹图谱分析方法可展现菌种水平差异(除了S.cerevisiae小于该遗传距离,大约为3),而H.burtonii、W.anomalus与S.fibuligera都展现了菌种内部基因型之间的差异,其中基因型10、11、12、15、16与17之间的遗传关系较近,而其余的菌种内部基因型之间的遗传距离都大于2,遗传关系相对较远。
2.3 种间和种内聚类分析比较
TRtRNA指纹图谱聚类分析(图8)重点体现了种内差异,菌种水平差异与26S rRNA基因D1/D2区域序列系统发育分析(图2)结果具有一定的一致性,如展现了S.malanga和S.fibuligera与W.anomalus、H.burtonii和S.cerevisiae相对较远的遗传距离,以及H.burtonii和S.cerevisiae相对较近的遗传距离,不同点在于基因型3(H.burtoniiFBKL2.8018)的种内遗传距离远于S.cerevisiae,而26S rRNA基因D1/D2区域序列系统发育分析虽然体现了FBKL2.8062与其他菌株之间的遗传差异,但其与S.cerevisiae的遗传差异大于种内差异。因此依赖于扩增片段分析的TRtRNA指纹图谱聚类分析相较于26S rRNA基因D1/D2区域序列系统发育分析而言,更适于展现种内差异,而依赖于序列分析的后者更适于展现菌种水平遗传差异。5.8S-ITS-RFLP分析虽然体现了与26S rRNA基因D1/D2区域序列系统发育分析类似的S.malanga和S.fibuligera较近的遗传距离(图5),但展现了S.cerevisiae与T.asahii较近而与其余菌种较远的遗传距离,这一点与依赖于序列分析的后者不同。因此,依赖于扩增片段大小分析的TRtRNA指纹图谱分析方法和5.8S-ITS-RFLP分析方法若能获取不同片段的序列信息,将增强其遗传距离聚类分析的能力。
3 讨论
酿酒小曲中的酵母菌在发酵中主要起酒化、酯化产香等功能,部分酵母菌可产生淀粉酶起糖化作用[1,3,23]。本研究所分离鉴定的6 种酵母菌作为潜在的小曲功能酵母,经课题组前期研究和文献查阅证实S.cerevisiae具有高产乙醇、低产高级醇的能力[20-21],W.anomalus具有高产乙酸乙酯等酯类能力[22-23],H.burtonii可产生大量酯类和醇类等风味物质[24-25],S.fibuligera具有产淀粉酶和酸性蛋白酶等酶的能力[26-28]。为贵州传统小曲中功能酵母菌的进一步开发利用提供分子身份,本研究在前期贵州传统小曲酵母菌分离培养的基础上,采用26S rRNA基因D1/D2区域序列分析和5.8S-ITS-RFLP分析方法确定酵母菌菌种水平分类地位和特征,并进一步采用TRtRNA指纹图谱法分析酵母菌的种内差异及基因型特征,鉴定的6 种酵母菌种有3 种展现了种内差异。首先,WL培养基作为常用酵母菌鉴定培养基共区分了8 种不同的菌落特征,但据目前报道的菌落特征库只能将S.cerevisiae初步确定,其余菌种无法确知属种名称。其次,26S rRNA基因D1/D2区域序列分析将59 株酵母菌鉴定到菌种水平,并展现了不同菌种之间的遗传距离和H.burtonii菌种内部的序列差异,而本研究首次拓展了除S.cerevisiae之外其余5 个酵母菌种的5.8S-ITS-RFLP特征图谱,可为后续快速检测菌种身份提供分子方法[29],虽然酶切聚类仅依赖4 个酶切图谱特征展开,但依然体现了与26S rRNA基因D1/D2区域序列分析相似的结论,即S.malanga和S.fibuligera菌种之间的遗传距离近于其他4 个酵母菌种,而T.asahii与酵母菌种之间的遗传距离较远。最终,本研究所采用的TRtRNA指纹图谱法由Barquet等[18]专门为体现非酵母属酵母菌种之间和种内差异所设计,在本研究TRtRNA指纹图谱法体现了59 株酵母菌之间的菌种水平之间和种内之间的分子差异,3 个菌种H.burtonii、W.anomalus与S.fibuligera都体现了较远遗传距离上(大于2小于3.6)的种内差异,说明TRtRNA指纹图谱法适用于某些酵母菌种的种内遗传多样性研究。贵州传统小曲中的主导优势菌种S.fibuligera的基因型12、14和17分别由分离自不同地区小曲的酵母菌组成,说明基因型分布可能与小曲地区来源关联较小。此外,Interdelta指纹图谱分析和微卫星分子标记法是常用的S.cerevisiae种内分型方法[18,30],而本研究中8 株S.cerevisiae未展现TRtRNA指纹图谱差异,8 株S.malanga菌株的TRtRNA指纹图谱单一,说明TRtRNA指纹图谱法可能无法在这两个酵母菌种中展现较为丰富基因型差异,但也可能因本研究中这两个菌种分离自同地区小曲因此基因型差异较少。
4 结论
从贵州5 个不同地区传统小曲中分离鉴定了6 个酵母菌种T.asahii(1 株)、S.fibuligera(32 株)、S.malanga(8 株)、H.burtonii(4 株)、W.anomalus(6 株)和S.cerevisiae(8 株),展示了6 个酵母菌种的WL培养基菌落特征、26S rRNA基因D1/D2区域序列系统发育特征、5.8S-ITS-RFLP酶切图谱特征和TRtRNA指纹图谱特征,尤其是TRtRNA指纹图谱特征展现了H.burtonii、W.anomalus与S.fibuligera的种内差异,为课题组进一步研究其低产高级醇和酯化产香等功能提供了可追踪的分子身份。
猜你喜欢
中学生学习报(2022年23期)2022-05-18
昆明医科大学学报(2022年1期)2022-02-28
军事文摘(2021年18期)2021-12-02
军事文摘·科学少年(2021年1期)2021-02-04
当代水产(2019年11期)2019-12-23
当代水产(2019年3期)2019-05-14
天然产物研究与开发(2018年9期)2018-10-08
机械设计与制造工程(2018年8期)2018-09-01
黄河之声(2016年12期)2016-11-07
新作文·初中版(2016年10期)2016-11-05
