
运动调节线粒体-内质网结构偶联缓解衰老性肌萎缩的研究进展*
2023-02-07 08:35高久翔
中国病理生理杂志 2023年1期
高久翔, 于 亮
(北京体育大学,北京 100084)
随着人口老龄化的加剧,衰老导致骨骼肌组织数量和质量退化,造成肌肉萎缩的发生率日趋增加,威胁着老年人的健康。衰老性肌萎缩(即肌肉减少症,sarcopenia)是一种慢性退行性疾病,主要表征为肌肉质量、力量和功能的下降,伴随摔倒、致残、发病和死亡率的增加[1],可能与线粒体功能障碍、内质网应激(endoplasmic reticulum stress, ERS)等反应有关[2-3]。预防衰老性肌萎缩将成为老年医学领域亟待解决的重要问题,运动作为一种经济有效的健康促进手段,已被证明对缓解衰老性肌萎缩具有一定意义[1]。
线粒体-内质网结构偶联(即线粒体相关内质网膜,mitochondria-associated endoplasmic reticulum membranes, MAMs)是线粒体和内质网间高度动态紧密连接的结构偶联域,对实现真核细胞内线粒体和内质网之间信号的快速传导与转换,维持细胞正常生命活动有着重要的意义。但MAMs在衰老性肌萎缩中发挥的作用机制仍未阐明,鉴于此本文就MAMs与衰老性肌萎缩的关系进行梳理,并阐述运动通过调节MAMs缓解衰老性肌萎缩发生的可能机制,为MAMs成为预防衰老性肌萎缩的靶点提供理论参考。
1 MAMs
MAMs是一个重要的亚显微结构,于20世纪90年代通过细胞片段化分离技术被发现并确认,富含磷脂和鞘糖脂合成酶,以及伴侣蛋白,控制蛋白质转运以及钙信号和其他代谢物在这两个细胞器间的传递,维持细胞的生物能量学和完整性。线粒体和内质网膜间某些区域重叠的部位(线粒体外膜5%~20%)彼此“连接”,但又未发生膜融合(距离约10~30 nm)[4],虽然这些接触只涉及膜表面的很小部分,但对细胞内通信起着重要的介导作用[5]。目前认为MAMs参与维持线粒体和内质网的正常功能,与细胞Ca2+稳态、氧化应激、线粒体动力学、ERS、脂质代谢、炎症、凋亡、自噬等密切相关[6]。
酵母细胞中,内质网-线粒体接触结构(ER-mitochondria encounter structures, ERMES)是包括线粒体外膜蛋白(Mdm10和Mdm34)、胞质蛋白(Mdm12)和整合内质网蛋白(Mmm1)的异源四聚体蛋白复合物,允许可溶性脂质载体蛋白进行有效的脂质转运[4],线粒体对ERMES结构完整性敏感,线粒体会因其组件的缺失而造成片段化。哺乳动物细胞中,组成MAMs结构的蛋白众多且复杂,目前已发现1 300多种蛋白富集在 MAMs中[7]共同参与、维持、调节MAMs活性,根据细胞功能主要可分为8类,见表1。
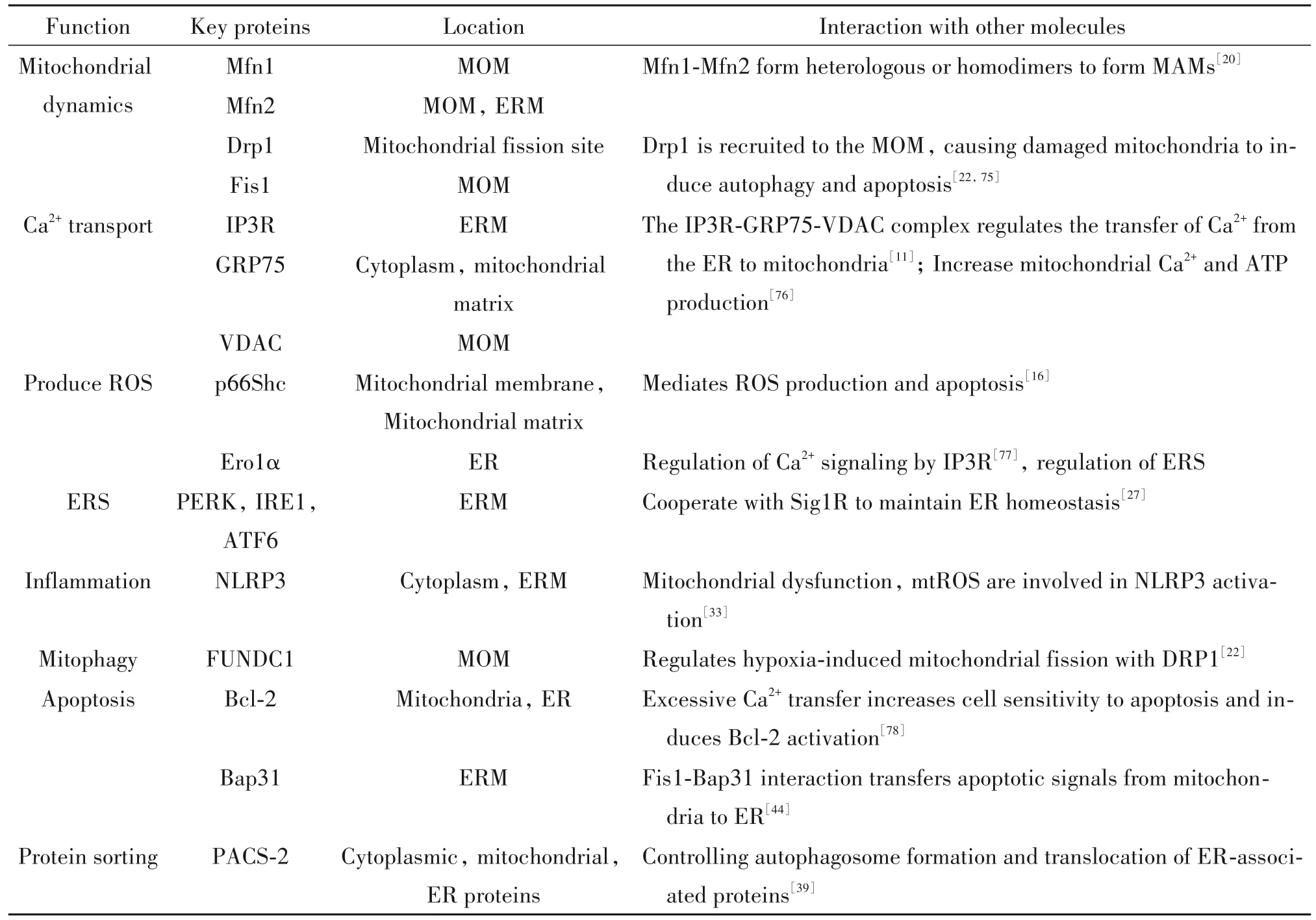
表1 MAMs中调节细胞功能的主要蛋白(哺乳动物细胞)Table 1. Major proteins that regulate cell function in MAMs (mammalian cells)
2 MAMs功能与衰老性肌萎缩
衰老导致的肌萎缩常伴有骨骼肌质量、力量、耐力、代谢水平、肌纤维数量及横截面积下降,脂肪及结缔组织增多,其本质特征是骨骼肌蛋白质合成水平降低,降解水平增强,致净蛋白平衡(net muscle protein balance, NMPB)紊乱。线粒体功能紊乱和ERS会造成NMPB,可概括为:(1)衰老增加线粒体活性氧(reactive oxygen species, ROS)水平,降低DNA修复能力,诱发慢性炎症、细胞凋亡、细胞自噬等生理过程破坏NMPB,致肌肉萎缩;(2)衰老导致氧化损伤增加,内质网分子伴侣活性和表达水平下降、内质网适应能力降低激活ERS途径,并启动内质网未折叠蛋白反应(endoplasmic reticulum unfolded protein response, UPRER),抑制真核细胞翻译起始因子2α(eukaryotic translation initiation factor 2α, eIF2α),造成骨骼肌合成代谢抵抗,破坏NMPB加剧肌萎缩的发生[2]。此外,UPRER与线粒体动力学、凋亡、自噬等途径关系密切[8],提示线粒体和内质网之间可能存在特定结构域,在衰老性肌萎缩中协同发挥相应的生理功能。MAMs由线粒体外膜、内质网膜及其亚结构域和一系列连接蛋白组成,是线粒体和内质网间形成的结构偶联,介导两细胞器间信号传导,为线粒体和内质网参与生物活性和调节细胞功能提供场所。因此,推测MAMs可能是衰老性肌萎缩发生的场所之一。
2.1 MAMs-Ca2+转移-衰老性肌萎缩 内质网和线粒体是参与Ca2+稳态调控的重要细胞器,Ca2+摄取对线粒体内在功能及代谢活动的调节至关重要[9],MAMs的状态和效率是决定Ca2+从内质网向线粒体转移的重要调控因素[10]。内质网中Ca2+通过 1,4,5-三磷酸肌醇受体(inositol 1,4,5-triphosphate receptor, IP3R)释放,经线粒体外膜电压依赖性阴离子通道(voltagedependent anion channel, VDAC)转移至线粒体,与葡萄 糖 调 节 蛋 白 75(glucose-regulated protein 75,GRP75)形成介导Ca2+从内质网转移到线粒体的IP3R-GRP75-VDAC多蛋白复合物[11]。线粒体钙单向转运体(mitochondrial calcium uniporter, MCU)负责感知Ca2+浓度,当Ca2+浓度较低,对Ca2+亲和力下降,阻止Ca2+摄入线粒体,相反MAMs中出现高钙浓度时,确保MCU激活和线粒体内Ca2+的迅速转移。
衰老过程中线粒体Ca2+失调导致MCU复合物功能受损以及线粒体代谢和动力学异常[12]。研究报道,MCU和其调节因子钙摄取家族成员1(mitochondrial calcium uptake 1, MICU1)的敲除导致骨骼肌萎缩[13]。钙摄取家族成员 3(mitochondrial calcium uptake 3, MICU3)是神经系统中MCU调节因子,其通过与MICU1合作增加线粒体Ca2+摄取[14]。Yang等[15]发现,在D-Gal诱导C2C12小鼠成肌细胞衰老模型中,MICU3的表达下调导致线粒体Ca2+摄取显著减少,MICU3过表达促进线粒体Ca2+稳态和功能,减弱氧化应激和细胞凋亡,恢复骨骼肌质量和功能。因此,可推测衰老性肌萎缩与MAMs结构中MCU及其调节因子功能减退,导致Ca2+通过IP3R-GRP75-VDAC复合物转移时受阻有关。
2.2 MAMs-氧化应激-衰老性肌萎缩 MAMs与氧化应激的联系主要表现在ROS的生成。内质网氧化还 原 蛋 白 1α(endoplasmic reticulum oxidoreductin 1α, Ero1α)包括α和β亚型,约75%位于MAMs,控制内质网氧化还原稳态,调控ERS,其表达水平直接与ROS的产生相关。信号衔接蛋白(66 kD isoform of the growth factor adapter Shc, p66Shc)定位于线粒体外膜、线粒体内外膜间及线粒体基质,其表达随年龄的增长而增加。氧化应激时,p66Shc被蛋白激酶Cβ(protein kinase Cβ, PKCβ)诱导并在丝氨酸36位点磷酸化,继而转移至线粒体或MAMs,介导ROS的产生和凋亡信号通路[16]。此外,ROS的产生促进内质网中的Ca2+通过MAMs向线粒体转移,致线粒体Ca2+储备、去极化、氧化磷酸化异常,造成线粒体电子传递链与呼吸复合物Ⅰ和Ⅲ解偶联,加剧线粒体ROS(mtROS)的产生[17]。
随年龄增长,线粒体和NADPH氧化酶会产生更多的ROS[18],尽管肌肉中的抗氧化酶活性随年龄的增长而增加,但这种补偿性适应不足以抵消氧化应激水平的上升。衰老骨骼肌中,氧化应激诱导线粒体DNA(mtDNA)突变并产生ROS,进一步导致电子传递链(electron transport chain , ETC)组件缺陷,缺陷的亚基进入ETC破坏氧化磷酸化,减少ATP合成及线粒体呼吸,并进一步增加ROS的产生,线粒体ROS的积累引发“恶性循环”导致组织退化、骨骼肌萎缩、肌肉功能障碍和纤维组织增加。熊建团等[19]发现衰老骨骼肌组织中Ero1α蛋白表达下降,启动子区DNA甲基化水平提高,同时骨骼肌细胞出现凋亡。可见,ROS及相关氧化损伤或氧化还原信号缺陷是诱发肌萎缩的原因之一,衰老引起的mtDNA突变、mtROS产生和内质网Ero1α DNA高甲基化是肌肉减少症的潜在原因。未来在对衰老性肌萎缩深入研究时也可考虑Ca2+转移与氧化应激间的关系。
2.3 MAMs-线粒体动力学失衡-衰老性肌萎缩 线粒体动力学指线粒体通过不断融合与分裂保持动态平衡的过程。线粒体融合和分裂蛋白主要集中于MAMs,是优化线粒体功能和质量控制的关键。介导线粒体融合的蛋白主要为定位于线粒体外膜的线粒体融合蛋白1/2(mitofusin 1/2, Mfn1/2),二者形成同型或异型二聚体复合物共同调控MAMs的结构和功能,并抑制内质网和线粒体的接近,促进线粒体外膜融合。Mfn2在MAMs也有定位,表明MAMs可能参与Mfn2依赖的线粒体融合[20]。线粒体内膜视神经萎缩蛋白1(optic atrophy 1 protein, OPA1)同Mfn1共同参与线粒体内膜融合。应激状态下线粒体膜电位降低并发生片段化,募集发动蛋白相关蛋白1(dynamin-related protein 1, Drp1)被其两个衔接蛋白线粒体分裂因子(mitochondrial fission factor, Mff)和线粒体分裂蛋白1(mitochondrial fission protein 1, Fis1)招募至MAMs处[21]。此外,线粒体外膜FUN14结构域蛋白1(FUN14 domain containing 1, FUNDC1)与钙联蛋白(calnexin)和Drp1相互作用并积聚于MAMs上,调节低氧诱导的线粒体分裂[22]。
衰老过程中线粒体动力学失衡导致线粒体形态改变及功能障碍,表现为:线粒体体积增大和嵴的破坏,ROS致mtDNA氧化损伤增加,氧化能力的减弱,进而导致增龄性骨骼肌的减少、肌肉疲劳、肌肉耐力下降以及力量的减弱。Chen等[23]报道了Mfn1和Mfn2的缺失导致mtDNA突变,而突变的累积导致线粒体功能障碍和肌肉萎缩。Romanello等观察到Drp1和Fis1的过度表达触发了线粒体片段化和功能障碍,激活线粒体自噬,导致肌纤维萎缩,对骨骼肌中Fis1和Drp1的基因沉默抑制线粒体分裂,可防止肌肉萎缩[24]。可见,线粒体融合有益于维持骨骼肌健康,线粒体过度分裂是肌肉衰老的重要原因。但目前就线粒体动力学与MAMs其他功能间的联系尚待确认。
2.4 MAMs-ERS-衰老性肌萎缩 ERS是细胞为应对内质网腔内错误折叠或未折叠蛋白及Ca2+紊乱等情况,通过蛋白激酶样内质网激酶(protein kinase-1ike ER kinase, PERK)、肌醇必需激酶1(inositol-requiring protein 1, IRE1)和激活转录因子6(activating transcription factor 6, ATF6)3种膜蛋白触发UPRER及凋亡等通路的反应过程,以恢复和维持内质网稳态[25]。MAMs参与 ERS主要表现在:Mfn2通过与PERK相互作用,降低PERK的表达,PERK通过其下游靶点活化转录因子4(activating transcription factor 4, ATF4)对C/EBP同源蛋白(C/EBP homologous protein, CHOP)和促凋亡因子Bax/Bak进行调控[26]。协同钙调蛋白Sigma 1受体(Sigma 1 receptor, Sig1R)通过PERK-eIF2α-ATF4途径致其表达上调维持内质网稳态[27];IRE1α 通过线粒体 Ca2+超载诱导 细 胞死亡[28]。
适宜的UPRER可一定程度提高内质网折叠蛋白能力,缓解蛋白质折叠负荷,重塑内质网稳态,缓解衰老性肌萎缩,过度激活的UPRER可能加剧衰老性肌萎缩的发生[3]。Laura研究证实[29],老年功能依赖性人群相比老年独立人群,其肌肉质量、功能和蛋白合成能力下降,这种下降通过UPRER得到补偿,表现为IRE1α和ATF6显著提高,同时阻断自噬途径,UPRER随衰老可能出现受损及相关伴侣蛋白的活性下降,当UPRER无法缓解ERS时易导致UPRER过度激活,通过PERK-ATF4等途径激活CHOP的表达,诱导促凋亡因子Bax/Bak转位,致萎缩因子MuRF1和MAFbx表达上调,加剧骨骼肌质量与力量的丢失[30]。可见,机体通过UPRER应对ERS的能力有限,衰老进程中适宜的UPRER可一定程度缓解肌萎缩的发生,但衰老导致过度激活UPRER时,机体可能通过介导细胞凋亡加剧肌萎缩的发生。
2.5 MAMs-炎症反应-衰老性肌萎缩 线粒体和内质网“cross talk”会影响炎症小体的形成和调节,NLR家族含pyrin域蛋白3(NLR family pyrin domain containing 3, NLRP3)是唯一与MAMs相关的炎症复合体,可被ROS激活[31]。NLRP3通常位于细胞质和内质网膜,炎症激活时,ROS将NLRP3及凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing a CARD, ASC)募集到MAMs感知线粒体损伤[32],可能由于线粒体功能障碍和氧化的mtDNA产生mtROS参与NLRP3激活[33]。硫氧还蛋白相互作用蛋白(thioredoxin-interacting protein,TXNIP)可直接与NLRP3结合,在氧化应激或接受到NLRP3炎症小体激活信号时,TXNIP可从细胞核转位到线粒体或MAMs,ERS时内质网PERK和IRE1α通路激活TXNIP[34]。通过将 MAMs结构成分 VDAC1/2 敲除,发现NLRP3炎症小体的形成受影响,证实VDAC介导并参与炎症反应过程[31]。
近年发现NLRP3参与炎症反应及衰老所致的肌萎缩,McBride等报道[35]NLRP3炎症小体与老年小鼠肌纤维数量下降有密切关系,NLRP3缺失的小鼠肌肉力量和耐力相对增加,且肌纤维数量的增龄性下调现象亦得到抑制。前文所述,衰老可致ERS并激活PERK和IRE1α通路致肌萎缩,而是否进一步激活TXNIP在衰老性肌萎缩相关研究中尚不清楚,通过各分子间相互关系,我们推测衰老致ERS可通过PERK/IRE1α-TXNIP与NLRP3直接结合致肌萎缩。此外,衰老导致MCU下调致VDAC复合物无法正常转移Ca2+,但研究尚未发现不同月龄小鼠VDAC蛋白和mRNA表达的变化[36]。推测,衰老性肌萎缩中的NLRP3炎症小体的形成可能亦是VDAC参与所致。因此,衰老可能与TXNIP、VDAC等通过NLRP3介导炎症反应致骨骼肌萎缩有关。
2.6 MAMs-自噬-衰老性肌萎缩 自噬是真核生物细胞内通过形成自噬小体以完成自我消化的过程,研究发现自噬体的双层膜结构源于内质网,通过MAMs发生转移[37]。目前认为与自噬相关的MAMs蛋白或信号通路主要有:自噬相关蛋白14(autophagy-related gene 14, Atg14)、自噬相关蛋白5(autophagy-related gene 5, Atg5)、哺乳动物雷帕霉素受体复合 物 2(mammalian target of rapamycin complex 2,mTORC2)、PTEN 诱导的激酶 1(PTEN-induced kinase 1, PINK1)/parkin 信号通路、Bcl-2/腺病毒 E1B 19 kD相互作用蛋白3(adenovirus E1B 19 kD interacting protein 3,BNIP3L)途径和FUNDC1途径。饥饿状态下MAMs结构的破坏可阻止Atg14和Atg5形成自噬体[38]。自噬关键分子mTORC2激活蛋白激酶B(protein kinase B, PKB/Akt),二者共同调控IP3R3磷酸化和Ca2+释放。MAMs相关蛋白Mfn2和磷酸呋喃酸分簇蛋白2(phosphofurin acidic cluster sorting protein-2,PACS-2)敲除,限制自噬体的形成及相关内质网相关蛋白的转位[39]。CCCP治疗后,PINK1和parkin在MAMs中聚集,调节线粒体和内质网间的相互作用[40]。缺氧条件下,FUNDC1充当线粒体自噬受体,募集自噬体致线粒体降解,也可招募Drp1驱动线粒体分裂,在MAMs处整合线粒体分裂和线粒体自噬以应对缺氧[22]。以上均可证实MAMs蛋白和自噬之间的功能联系密切。
衰老进程中细胞自噬水平下降,导致无法清除错误折叠的蛋白和受损的细胞器可能是诱导衰老性肌萎缩的原因。研究发现线粒体自噬蛋白Bnip3和parkin的表达会随着年龄的增长而降低[41]。24月龄小数小鼠股四头肌自噬相关蛋白LC3水平显著降低,并伴有肌纤维萎缩及核聚集现象[42]。但Romanello等[24]表明Bnip3过表达诱导线粒体断裂、自噬水平增高并引起肌肉萎缩。以上研究提示,衰老性肌萎缩与MAMs处自噬缺陷或功能障碍高度相关,衰老致自噬的不足和过度表达均会导致不同程度肌肉萎缩。
2.7 MAMs-凋亡-衰老性肌萎缩 线粒体和内质网交换时Ca2+超载是促进凋亡发生的重要信号。Ca2+从内质网到线粒体过度转移触发线粒体通透性转换孔(mitochondrial permeability transition pores, mPTP)开放,导致线粒体内膜通透化、ATP合成停止、线粒体肿胀、外膜破裂、细胞色素c释放,通过与IP3R结合促进凋亡发生[43]。B细胞受体相关蛋白31(B-cell receptor-associated protein 31, Bap31)通过与 Fis1结合成复合物,募集并激活procaspase-8,将Bap31切割成p20Bap31促凋亡形式,刺激内质网Ca2+释放,致线粒体 Ca2+超载、进一步开放 mPTP 促进凋亡[44]。此外,凋亡时DRP1刺激Bax寡聚化增加[45],且Mfn2与Bax在凋亡过程中存在共定位情况。
衰老性肌萎缩发生的机制与肌肉细胞凋亡和再生不平衡有关,动物实验观察到33月龄雄性FBN大鼠较9月龄大鼠跖肌质量降低了22%,Bax蛋白表达及凋亡指数增加。细胞内或细胞质Ca2+的持续升高是细胞凋亡导致肌肉蛋白降解肌肉功能受损的关键,钙蛋白酶介导的肌肉萎缩与细胞内钙离子升高或Ca2+过载密切相关[46]。综上,衰老细胞凋亡相关蛋白表达增高,同时MAMs处Ca2+超载是导致细胞凋亡是诱发肌萎缩的重要原因,但Ca2+经何种通路诱发凋亡尚不明晰。
对上述已有文献总结发现,衰老性肌萎缩的发生机制可能与MAMs处Ca2+转移异常、氧化应激、线粒体动力学失衡、ERS等有关,伴有炎症、自噬、凋亡等现象。衰老诱发的肌萎缩机制由多因素共同决定,炎症反应的发生可由ERS和氧化应激所致,自噬(线粒体)与线粒体动力学之间存在关系,凋亡可由过度ERS和Ca2+转移异常激活(图1)。
3 运动通过调节MAMs缓解衰老性肌萎缩的潜在分子机制
衰老进程中骨骼肌丢失的可能内在因素与线粒体和内质网功能障碍有关。骨骼肌长期对运动的适应性应答是增加或维持肌肉质量、力量、耐力的关键,但目前对于运动防护肌萎缩的研究多单独从调控线粒体自噬或减轻ERS角度进行论述[47],实际过程中二者往往联合发挥生理功能以进行稳态调控。因此,深入了解运动如何联合调控线粒体和内质网缓解衰老性肌萎缩更为有意义。本文依次从线粒体、内质网及二者间的区域,分析运动如何调节MAMs功能缓解衰老性肌萎缩,并对有氧、抗阻和复合运动方式调控MAMs功能缓解衰老性肌萎缩进行梳理,意在阐述运动调节MAMs缓解衰老性肌萎缩的潜在分子机制。
3.1 运动通过调节MAMs功能缓解衰老性肌萎缩
3.1.1 运动调节线粒体功能与衰老性肌萎缩 随年龄的增长,线粒体动力学效率降低,生物合成减少,导致自噬、凋亡水平异常,发生线粒体融合和分裂失衡。适度的运动是加速降解衰老和损伤的线粒体,促进骨骼肌线粒体生物合成,改善骨骼肌线粒体动力学系统,维持细胞健康的重要途径。
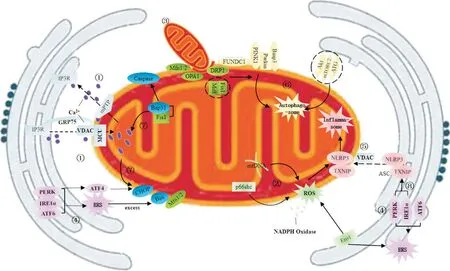
Figure 1. The relationship between sarcopenia and MAMs function. Process ①: Ca2+ transfer (uptake/release); ②: oxidative stress;③: mitochondrial dynamics (fusion/fission); ④: endoplasmic reticulum stress (ERS); ⑤: inflammatory response; ⑥:autophagy; ⑦: apoptosis; ⑧: inferred path.图1 衰老性肌萎缩与MAMs功能关系
长期有氧运动调控衰老肌细胞线粒体动力学可通过提高线粒体氧化磷酸化及肌细胞中线粒体Mfn2和Drp1基因表达水平,改善线粒体融合和分裂过程,维持更高水平线粒体的平衡。此外,运动中产生的ROS可激活线粒体自噬,诱导Drp1和PINK1活化并靶向清除功能受损的线粒体,促进老年人骨骼肌内自噬标志物beclin-1、Atg7和p62及线粒体自噬标志物Bnip3和parkin水平的增加[48]。然而,由于老年人对运动适应性水平降低,过度的运动反而破坏线粒体结构和功能,进而产生更多ROS进一步加剧线粒体损伤,严重时甚至发生凋亡[49]。综上可知,运动可通过调控线粒体氧化应激、动力学、自噬及凋亡等途径促进骨骼肌MAMs功能重塑,诱导骨骼肌产生适应性改变,预防衰老性肌萎缩。
3.1.2 运动调节ERS与衰老性肌萎缩 内质网是蛋白质折叠和翻译后修饰发生的主要位点,应激条件下,错误或未折叠的蛋白在内质网中积累破坏内质网的稳态。衰老进程中氧化损伤增加、内质网分子伴侣活性和表达下降、内质网适应能力降低导致ERS进而激活UPRER。UPRER可直接对程度较轻的ERS发挥作用以恢复内质网稳态,但超过UPRER削弱ERS的范围时,持续的ERS加剧UPRER激活,进而介导JNK/IRE1α或NF-κB等凋亡通路,导致萎缩因子MuRF1和MAFbx表达上调,加剧骨骼肌质量与力量的丢失。
适宜的运动可通过激活或优化UPRER,削弱ERS缓解衰老性肌萎缩[3]。健康老年男性和女性在进行为期8周的抗阻运动后,UPRER三条信号轴PREK/IRE1/ATF4被激活[50]。需要注意,急性抗阻运动后24~48 h老年人骨骼肌BiP和ATF6 mRNA表达显著性升高[51],长期耐力或抗阻运动后未发现上述现象,这可能与以上三条轴在激活和失活时间上存在差异有关。另外,5周高强度有氧运动(坡度10°,跑速34 m/min,60 min)相比低强度有氧运动(坡度10°,跑速20 m/min,60 min)更能促进大鼠骨骼肌PGC-1α的表达,并降低骨骼肌ERS和凋亡信号,说明UPRER的激活可能与线粒体生物发生标记物PGC-1α表达水平有关[52]。以上提示,运动通过UPRER调节ERS延缓衰老性肌萎缩时需要注意运动方式、运动强度和运动时间等因素。此外,运动调节ERS过程中线粒体同时发挥重要的作用。
3.1.3 运动调节MAMs功能与衰老性肌萎缩 线粒体和内质网之间的区域在衰老性肌萎缩中同样发挥重要的功能,如Ca2+和炎症小体从内质网向线粒体转运均通过MAMs。此外,线粒体自噬过程中PINK1和parkin介导Mfn2等线粒体外膜蛋白泛素化,增加内质网和线粒体间的接触距离以促进线粒体降解[53]。
大鼠进行19个月转轮自由跑后,老年阶段炎性因子水平下降[54]。小鼠在进行45 min跑速为12 m/min耐力跑后,发育及DNA损伤反应调节基因1(regulated in development and DNA damage 1, REDD1)mRNA含量提升110%~500%;以12 m/min~20 m/min的递增跑速进行45 min后,REDD1mRNA含量提升170%~880%[55],REDD1可与MAMs上相关成分蛋白相互作用(如GRP75,调节Ca2+转运组件之一),干扰内质网和线粒体之间的相互作用[56],并且该现象与运动强度存在一定相关性。然而,大负荷运动会导致Ca2+从内质网向线粒体转移,表现在骨骼肌内质网中Ca2+浓度下降,线粒体中Ca2+浓度升高。多年运动经验的老年人,其肌肉内单位体积释放Ca2+单元高于无运动经验的老人[48,57]。运动降低内质网与线粒体之间的相互作用,增加二者之间的Ca2+转运现象看似矛盾,推测可能与运动通过改变线粒体和内质网膜之间的距离,使离子转运效率更高,但目前对于运动与线粒体和内质网膜间距的相关研究较少,还有待验证。
3.2 不同运动方式调节MAMs缓解衰老性肌萎缩的分子机制
3.2.1 有氧运动调节MAMs缓解衰老性肌萎缩的分子机制 适当规律的有氧运动不仅可以维持老年人心血管、骨骼和肌肉的健康,同时也是刺激骨骼肌中线粒体、内质网等细胞器适应肌肉对能量代谢的需求,缓解衰老性肌萎缩的重要方式。目前涉及有氧运动调节MAMs缓解衰老性肌萎缩的分子机制较为分散。人体实验研究表明,有多年骑行经历的老年人[58-59],腿部肌肉内线粒体融合蛋白Mfn1/2,线粒体分裂蛋白Fis1和Drp1的表达均明显高于无训练习惯老年人。Balan等[59]发现每周6 h以上骑行习惯的老人mtDNA/nDNA比值升高,这一结果与Kang等[60]和 Koltai等[61]的结果相似。Konopka等[62]在对老年人进行 12周,每周进行 3~4次 20~45 min,强度在60%~80%心率储备的自行车训练后发现,线粒体融合蛋白Mfn1/2,线粒体分裂蛋白Fis1表达升高,以上说明有氧强度的自行车运动对于减轻线粒体氧化应激,刺激线粒体融合与分裂具有一定意义。动物实验中发现衰老模型鼠长期进行有氧跑台、游泳和笼内转轮跑干预[61,63-64]后线粒体融合蛋白Mfn1/2,分裂蛋白Fis1和Drp1均出现不同程度下调,提示有氧运动对线粒体融合、分裂蛋白的调控表现出一致性,即“同升同降”以维持线粒体稳态平衡。刘文峰等[65]发现,为期10周、每周5 d、持续35 min、跑速20 m/min的耐力跑后,衰老大鼠IP3R及自噬相关蛋白Akt和mTOR表达升高,说明增强Ca2+转运和抑制因衰老导致的自噬功能下降是缓解衰老性肌萎缩的可能原因。此外也有研究得到[59,66-67]长期有氧干预后线粒体自噬相关蛋白PINK1和parkin表达上调,凋亡相关蛋白Bax/Bcl-2比值及caspase家族表达显著下调[66-70],说明有氧运动缓解衰老性肌萎缩与激活线粒体自噬,抑制凋亡高度相关。Belaya等[54]对24月龄大鼠进行19个月转轮自由跑发现,直接与NLRP3(唯一与MAMs相关的炎症复合体)结合的TXNIP表达下降,同时ERS蛋白GRP78表达下降,说明有氧运动可减轻炎症反应及ERS以缓解衰老性肌萎缩。然而,目前少有研究横向比较运动强度对MAMs功能的影响,并且动物实验操作过程中难以保证每只动物以其个体特定VO2max百分比强度进行运动干预。因此,探索更精准的运动剂量对MAMs功能的影响,为老年人预防肌萎缩提供最佳的锻炼模式可作为下阶段的研究方向。见表2。
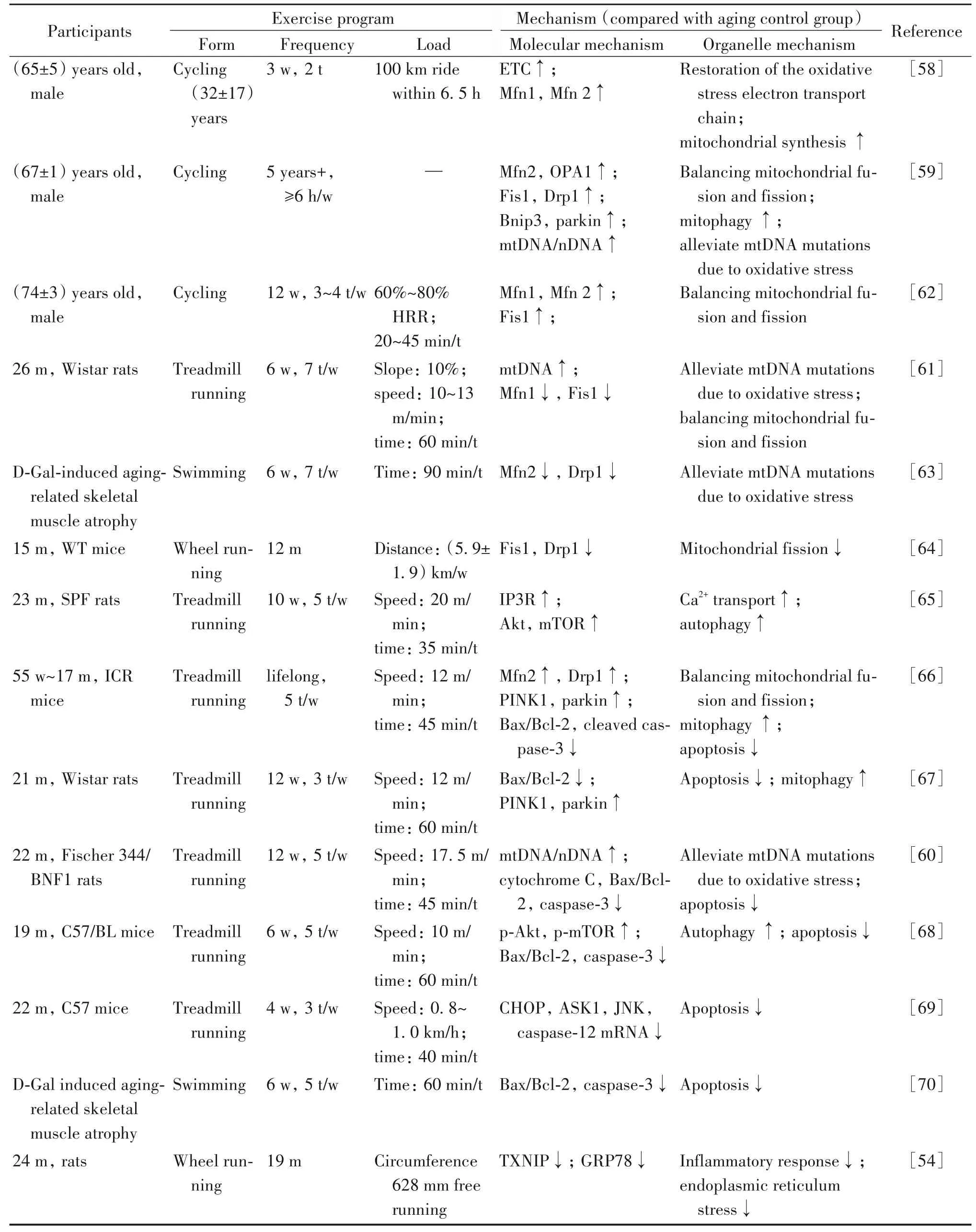
表2 有氧运动通过MAMs缓解衰老性肌萎缩的机制Table 2. Mechanisms of aerobic exercise attenuating sarcopenia through MAMs
3.2.2 抗阻运动调节MAMs缓解衰老性肌萎缩的分子机制 抗阻训练已被证实是一种改善老年人的运动表现并缓解肌肉力量下降的干预措施,老年人进行适当的抗阻运动可维持瘦体重、肌肉力量,提高平衡及协调性以保证完成相关功能性动作。目前对抗阻运动调节MAMs缓解衰老性肌萎缩的分子机制研究发现,老年男女受试者进行每周2次,每次3组,每组10~12次,负荷强度约在70%~90%的全身力量训练,10周后发现骨骼肌中线粒体融合蛋白Mfn1/2表达升高[71],Estébanez等[50]同样发现8周,每周2次,每次3组,每组8~12次,负荷强度40%~75% 1RM渐增力量训练后,老年男女受试者Mfn1蛋白升高,即抗阻训练可刺激老年人线粒体合成以缓解萎缩。Estébanez等[50]的研究中 UPRER相关指标 p-PERK、p-IRE1和p-ATF4显著上调,结合Smiles等[72]的结论,UPRER的激活可能与线粒体生物发生标志物PGC-1α表达水平有关,提示线粒体和内质网可能在抗阻运动中联合发挥作用以缓解肌萎缩的发生。动物实验中发现老年鼠每周3次,渐增负重强度的爬梯4~12周均可使Bax/Bcl-2比值、caspase家族、细胞色素C等凋亡相关指标均下调[67,69,73]。但在对自噬相关机制研究时发现,老年人进行为期8周的抗阻训练后,线粒体自噬蛋白PINK1、parkin和Bnip3/Nix未改变[50];老年大鼠进行9周负重爬梯后,Akt-mTOR磷酸化水平下降[73],12周负重爬梯后PINK1和parkin上调[67]。
以上研究除了受试种群和运动周期不同外,其差异主要在于人体实验的运动强度在40%~75% 1RM,动物实验的负荷分别为10%~90%和20%~60%自身体重,提示机体对自噬机制的感知取决于对运动刺激的适应程度。此外,3组实验中除自噬相关变化外,同时伴有线粒体合成增强,凋亡被抑制的情况,提示抗阻运动缓解衰老性肌萎缩的分子机制并不是由单一因素决定,而是受多因素调控。还有观点认为抗阻运动诱导肌肉适应与合成代谢相关胰岛素样生长因子1(insulin-like growth factor 1, IGF-1)有关,并通过IGF1-PI3K-Akt-mTOR通路介导[74]。总之,抗阻运动调控MAMs缓解衰老性肌萎缩的分子机制更可能是由多种因素共同决定,主要以合成代谢为主,表现在刺激线粒体合成,抑制细胞凋亡,并且线粒体和内质网可能联合发挥作用。见表3。

表3 抗阻运动通过MAMs缓解衰老性肌萎缩的机制Table 3. Mechanisms of resistance exercise attenuating sarcopenia through MAMs
3.2.3 复合运动调节MAMs缓解衰老性肌萎缩的分子机制 单纯进行有氧或力量训练会因其重复性操作使人失去兴趣,增加高龄人群受伤风险,另外由于老年人mTOR信号的缺乏进行力量训练的效果可能较差[41],老年人在实际生活中进行复合形式的体力活动更为常见,并且运动效果更有利于提高老年人的体成分和体适能。通过对有多年复合运动或规律运动的老年运动员进行肌肉活检检测,其肌肉单位面积内Ca2+释放单元,线粒体数量显著高于普通老年人,线粒体自噬蛋白Binp3和parkin表达相对较高[48,57]。21月龄雄性大鼠进行12周,每周3次17.5 m/min的跑台和3次20%~60%体重的渐增负重爬梯训练后,骨骼肌内凋亡水平下降,线粒体自噬水平提高[67]。说明长期进行复合形式运动可以缓解衰老性肌萎缩与Ca2+转运能力及线粒体活性并激活线粒体自噬。以上可解释骨骼肌受Ca2+释放单元三联体和线粒体结构和功能的控制提供Ca2+和ATP,衰老导致Ca2+释放单元和线粒体减少,降低二者间偶联频率,运动训练对衰老性骨骼肌Ca2+和线粒体产生良性适应以缓解萎缩发生[57]。此外,因有氧运动中PGC-1α通路和抗阻运动中Akt/mTOR通路在肌肉适应中存在不兼容的现象,建议实际训练时避免二者进行同期训练。见表4。
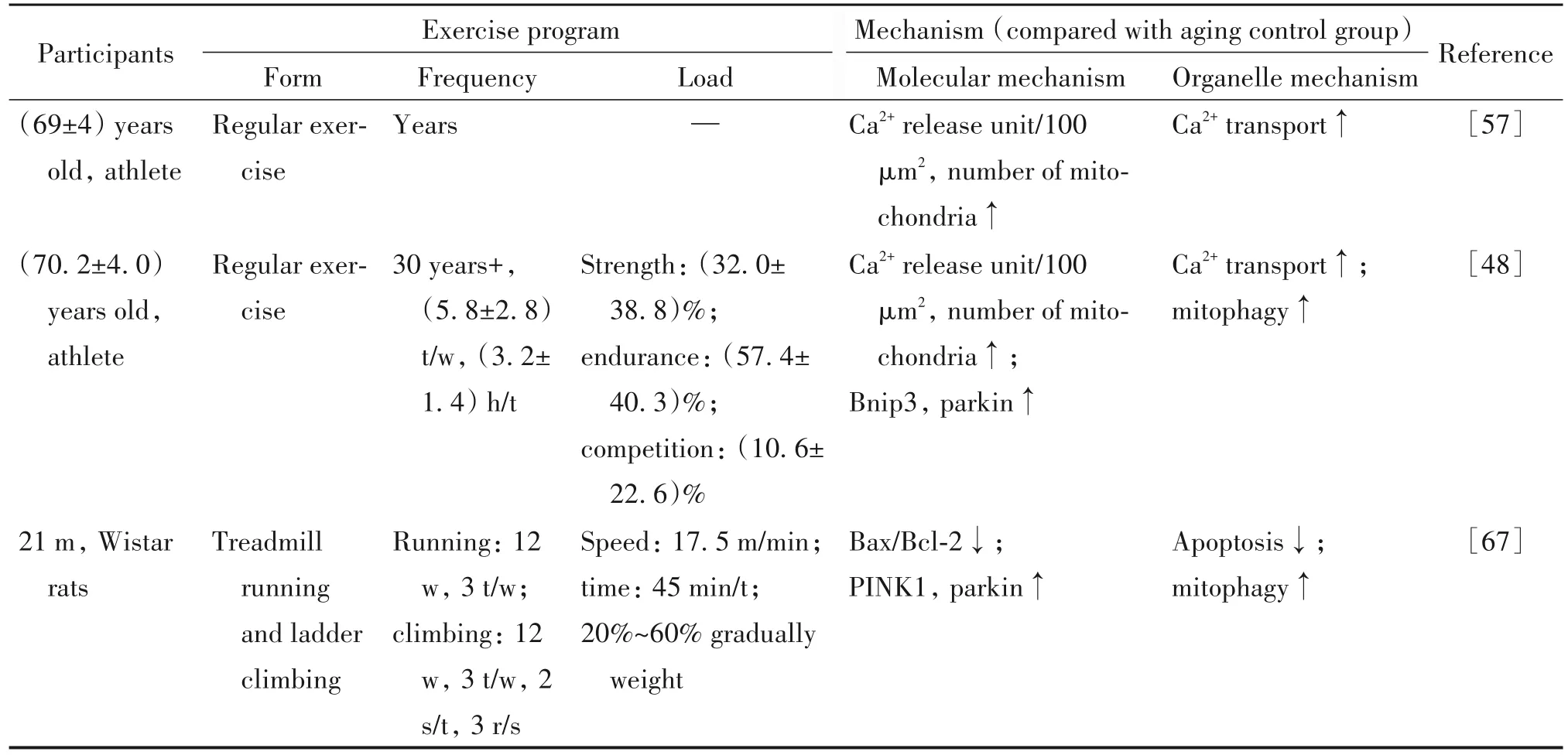
表4 复合运动通过MAMs缓解衰老性肌萎缩的机制Table 4. Mechanisms of comprehensive exercise attenuating sarcopenia through MAMs
4 小结与展望
衰老性肌萎缩是一种由多因素引起的不可避免的退行性生命现象,线粒体和内质网功能紊乱是其发生的因素之一。目前可认为衰老性肌萎缩的发生与MAMs处Ca2+转移异常、氧化应激、线粒体动力学失衡、ERS等有关,并伴有炎症、自噬、凋亡等现象。其中,炎症反应的发生可由ERS和氧化应激所致,自噬(线粒体)与线粒体动力学之间存在关系,凋亡可由过度ERS和Ca2+转移异常激活。运动作为重塑骨骼肌功能的有效方式,可靶向线粒体和内质网在MAMs处发挥重要作用。运动缓解衰老性肌萎缩的机制是由改善Ca2+转移,保证线粒体融合和分裂的平衡,缓解氧化应激及ERS,激活线粒体自噬,降低炎症和凋亡等因素共同决定。未来可以从以下几方面继续开展研究:(1)进一步挖掘MAMs中细胞功能,及诱使MAMs各组件联合发挥作用的枢纽分子,以便找到MAMs介导衰老性肌萎缩各反应之间的关联机制;(2)研究衰老对骨骼肌MAMs膜间距影响,运动是否通过改善膜间距提高各组件之间的传递效率缓解肌萎缩;(3)MAMs中TXNIP-NLRP3炎症通路与衰老性肌萎缩的直接证据尚不充分,MAMs各细胞功能与线粒体动力学之间的关系尚待进一步补充;(4)适度范围内研究不同训练方式、训练量、训练强度缓解肌萎缩的效果及对MAMs的调控情况,以期为指导老年人最佳的锻炼模式提供理论依据。
猜你喜欢
现代临床医学(2021年1期)2021-01-26
天然产物研究与开发(2018年2期)2018-04-04
安徽医科大学学报(2016年12期)2017-01-15
中国运动医学杂志(2016年3期)2016-07-10
中国运动医学杂志(2016年3期)2016-07-10
现代电生理学杂志(2016年1期)2016-07-10
中西医结合心脑血管病杂志(2016年20期)2016-03-01
医学研究杂志(2015年5期)2015-06-10
——疾病防治的新靶标
中国药理学通报(2015年3期)2015-06-09
中国康复理论与实践(2015年7期)2015-05-09
