
双调蛋白对支气管肺发育不良模型小鼠肺泡分化的影响*
2023-02-07 08:35尧惠慈卢红艳
中国病理生理杂志 2023年1期
尧惠慈, 卢红艳, 朱 玥, 乔 瑜, 季 玮
(江苏大学附属医院儿科, 江苏 镇江 212001)
支气管肺发育不良(bronchopulmonary dysplasia,BPD)是一种早产儿慢性肺部疾病,随着围产医学的进步,早产儿存活率提高,BPD的发病率也逐渐升高,其原因主要是发育中未成熟肺的损伤和修复之间的不平衡[1]。肺的发育主要通过上皮细胞和间充质细胞的相互作用介导,肺泡上皮由两种形态与功能不同的细胞组成:Ⅰ型肺泡上皮细胞(type I alveolar epithelial cell, AECI)和Ⅱ型肺泡上皮细胞(type II alveolar epithelial cell, AECII)。平足蛋白(podoplanin, T1α)是AECI的标志物[2],肺表面活性蛋白C(surfactant protein C, SP-C)是AECII的标志物[3],在肺上皮损伤正常修复中,AECII增殖并分化为AECI以促进肺泡结构和气血屏障形成,一旦出现分化障碍,肺损伤不可逆[4]。
目前认为BPD主要为肺血管和肺泡生长发育停滞[5]。长期暴露于85%高氧的新生鼠肺泡腔扩大,这表明存在肺发育受阻[6]。表皮生长因子家族成员双调蛋白(amphiregulin, AREG)在上皮细胞和间充质细胞中均表达,能调节不同类型细胞的增殖、凋亡和迁移,包括上皮细胞、成纤维细胞和免疫细胞[7]。有研究显示,AREG可促进气道上皮细胞和平滑肌细胞的增殖[8]。在呼吸机相关急性肺损伤模型中,肺组织内AREG表达上调,提示AREG表达升高可能参与呼吸机相关肺损伤[9]。在产前烟雾暴露诱导的BPD小鼠模型中,支气管和肺细胞受损可能与AREG-表皮生长因子受体(epidermal growth factor receptor, EGFR)信号转导有关[10]。EGFR是具有多种配体的酪氨酸激酶受体,在上皮细胞中,EGFR的极化及其下游信号的激活在定向细胞迁移中起重要作用[11]。目前,在高氧暴露所致BPD小鼠模型中,AREG对肺泡分化的影响尚不明确,本研究通过建立高氧暴露所致BPD模型小鼠,探讨AREG及EGFR在BPD小鼠肺组织中的动态表达及AREG表达变化对肺泡分化的影响,以期为研究BPD肺泡化障碍机制提供实验依据。
材料和方法
1 主要试剂
小鼠抗AREG多克隆抗体及重组小鼠AREG蛋白(recombinant AREG protein, rmAREG)购自R&D;兔抗EGFR多克隆抗体及小鼠抗β-actin单克隆抗体购自Cell Signaling Technology;兔抗SP-C多克隆抗体、山羊抗兔IgG及驴抗小鼠IgG均购自Abcam;小鼠抗T1α单克隆抗体购自Santa Cruz;HRP标记的山羊抗兔IgG及HRP标记的山羊抗小鼠IgG均购自上海碧云天生物技术有限公司;AREG慢病毒购自上海吉凯基因;PBS购自HyClone。
2 主要方法
2.1 模型制备 SPF级孕16~17 d健康C57BL/6小鼠由江苏大学实验动物中心提供[许可证号:SYXK(苏)2018-0053]。待小鼠出生后,将新生小鼠随机分为空气(normoxia, N)组和高氧(hyperoxia, H)组,利用高氧暴露复制小鼠BPD模型[12]。高氧组小鼠置于氧箱中,氧浓度为85%,空气组置于同一室内常压空气中;代母鼠每12 h在各组间更换一次,以避免氧中毒并排除不同组间代母鼠的影响,每日观察小鼠情况并记录。各组小鼠于空气或高氧暴露后7 d和14 d吸入麻醉处死并分为7 d空气(N7)组、7 d高氧(H7)组、14 d空气(N14)组和14 d高氧(H14)组。取出肺组织,左肺组织用于制作石蜡切片,观察免疫荧光检测,右肺组织冰冻保存用于相关蛋白检测。
2.2AREG过表达及敲低 取重组AREG蛋白5 μg溶于100 μL PBS中,于第10天给高氧暴露新生小鼠腹腔注射重组AREG蛋白(每只100 μL)实现AREG过表达[13],对照组于相同时间注射相同剂量PBS,并分为rmAREG组和PBS组,每组各5只。取小鼠AREG小干扰RNA(small interfering RNA, siRNA)慢病毒表达载体溶液10 μL(载体浓度为3×1011TU/L),于第10天给高氧暴露新生小鼠鼻内滴注,实现AREG敲低(AREG knockdown, KD)[14],对照组于相同时间滴注相同剂量阴性对照(negative control, NC)载体溶液,并分为NC组和KD组,每组各5只,各组小鼠于高氧第14天处死并进行后续实验。
2.3 Western blot检测肺组织中AREG、EGFR、SP-C及T1α的蛋白表达 RIPA裂解液(含PMSF、蛋白酶抑制剂)提取各组肺组织总蛋白,按每泳道10 μg总蛋白进行SDS-PAGE分离,湿式电转至PVDF膜上,5%脱脂奶粉溶液37 ℃封闭1 h后,分别加入小鼠抗AREG多克隆抗体(1∶500)、兔抗EGFR多克隆抗体、小鼠抗T1α单克隆抗体、兔抗SP-C多克隆抗体和小鼠抗β-actin单克隆抗体(均1∶1 000),4 ℃孵育过夜。洗膜,分别以HRP标记的山羊抗兔IgG和HRP标记的山羊抗小鼠IgG(均1∶5 000)孵育1 h,ECL化学发光显色。以目的蛋白与内参照β-actin蛋白条带的灰度值的比值表示目的蛋白相对表达水平。
2.4 免疫荧光双标技术检测SP-C/T1α表达 将肺组织切片脱蜡至水,抗原修复后用5% BSA在37 ℃封闭20 min,并与兔多克隆抗SP-C抗体(1∶100稀释)和小鼠单克隆抗T1α抗体(1∶100稀释)的混合物在4 ℃下孵育过夜,切片用PBS洗3次后,与山羊抗兔Ⅱ抗和驴抗小鼠Ⅱ抗(1∶1 000稀释)在37 ℃下避光孵育1 h,PBS洗3次后,采用DAPI室温避光染核,荧光倒置显微镜下观察肺组织的荧光染色,ImageJ软件分析阳性细胞数及共定位。
3 统计学处理
采用SPSS 11.0 统计软件对数据进行统计学分析,采用GraphPad 8.0软件绘制柱状图。计量资料以均数 ± 标准差(mean±SD)表示,两组间比较采用独立样本t检验,多组间比较采用单因素方差分析(one-way ANOVA),组间两两比较采用LSD法,以P<0.05 为差异有统计学意义。
结 果
1 高氧暴露BPD小鼠不同时点肺组织中AREG、EGFR、T1α及SP-C的蛋白表达情况
与同时点空气组相比,高氧组AREG、EGFR和T1α蛋白表达升高(P<0.01),SP-C蛋白表达水平显著降低(P<0.05)。在空气及高氧状态下,AREG及EGFR蛋白表达随时间呈上升趋势。见图1。
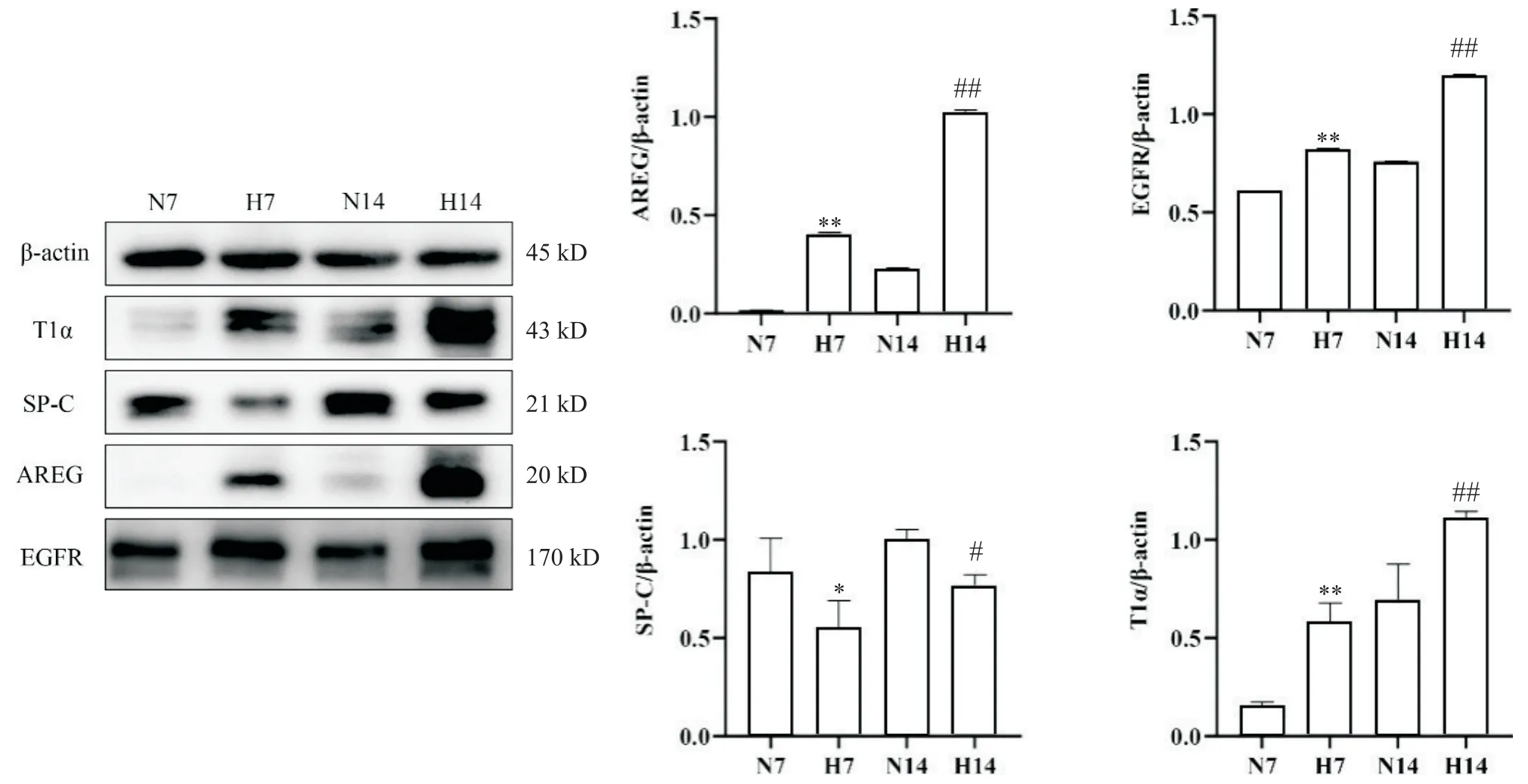
Figure 1. The protein expression of AREG, EGFR, SP-C and T1α in lung tissues of normoxia and hyperoxia mice at different time points. N7: normoxia for 7 d; H7: hyperoxia for 7 d; N14: normoxia for 14 d; H14: hyperoxia for 14 d. Mean±SD. n=5.*P<0.05, **P<0.01 vs N7 group; #P<0.05, ##P<0.01 vs N14 group.图1 空气组及高氧组不同时点小鼠肺组织中AREG、EGFR、SP-C及T1α的蛋白表达水平
2 高氧暴露BPD小鼠肺组织中SP-C和T1α免疫荧光共定位情况
采用肺上皮细胞标志物SP-C和T1α蛋白进行免疫荧光染色,绿色荧光为SP-C的阳性表达,红色荧光为T1α的阳性表达,蓝色荧光为DAPI标记的细胞核,SP-C和T1α双阳性则呈橙色。免疫荧光结果显示,相较于同时点空气组,高氧组中SP-C和T1α共定位细胞显著减少(P<0.05,P<0.01),见图2。
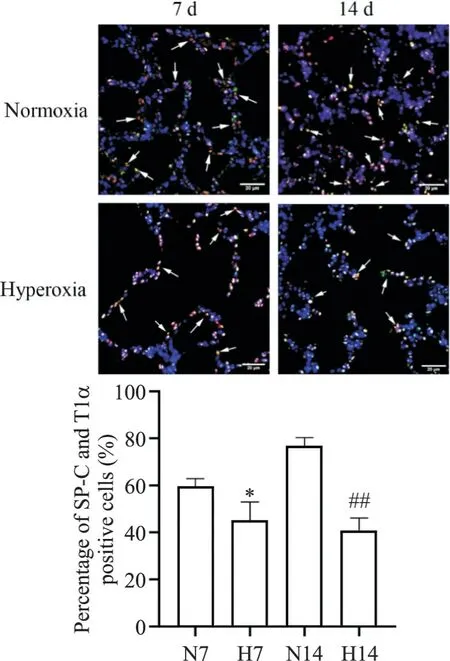
Figure 2. Immunofluorescence staining of SP-C (green) and T1α (red) in lung tissues of normoxia and hyperoxia mice at different time points (scale bar=20 μm).Blue indicates DAPI; arrows indicate SP-C and T1α positive cells. N7: normoxia for 7 d; H7: hyperoxia for 7 d; N14: normoxia for 14 d; H14: hyperoxia for 14 d. Mean±SD. n=5. *P<0.05 vs N7 group;**P<0.01 vs N14 group.图2 空气组及高氧组不同时点小鼠肺组织SP-C和T1α免疫荧光共定位情况
3 高氧暴露BPD小鼠过表达AREG后肺组织中AREG、EGFR、T1α及SP-C的蛋白表达情况
在高氧状态下,腹腔注射rmAREG后,与PBS组相比,rmAREG组中AREG和EGFR蛋白表达显著升高 (P<0.01),而SP-C和T1α蛋白表达显著降低(P<0.01),见图3。
4 高氧暴露BPD小鼠过表达AREG后肺组织中SP-C和T1α免疫荧光共定位情况
在高氧状态下,注射rmAREG后,rmAREG组中SP-C与T1α共定位细胞较PBS组显著减少(P<0.01),见图4。
5 高氧暴露BPD小鼠敲低AREG后肺组织中AREG、EGFR、T1α及SP-C的表达情况
在高氧状态下,小鼠鼻内滴注AREG siRNA慢病毒表达载体溶液后,与NC组比较,KD组中AREG和EGFR蛋白表达显著降低 (P<0.01),而SP-C和T1α蛋白表达则显著升高(P<0.01),见图5。
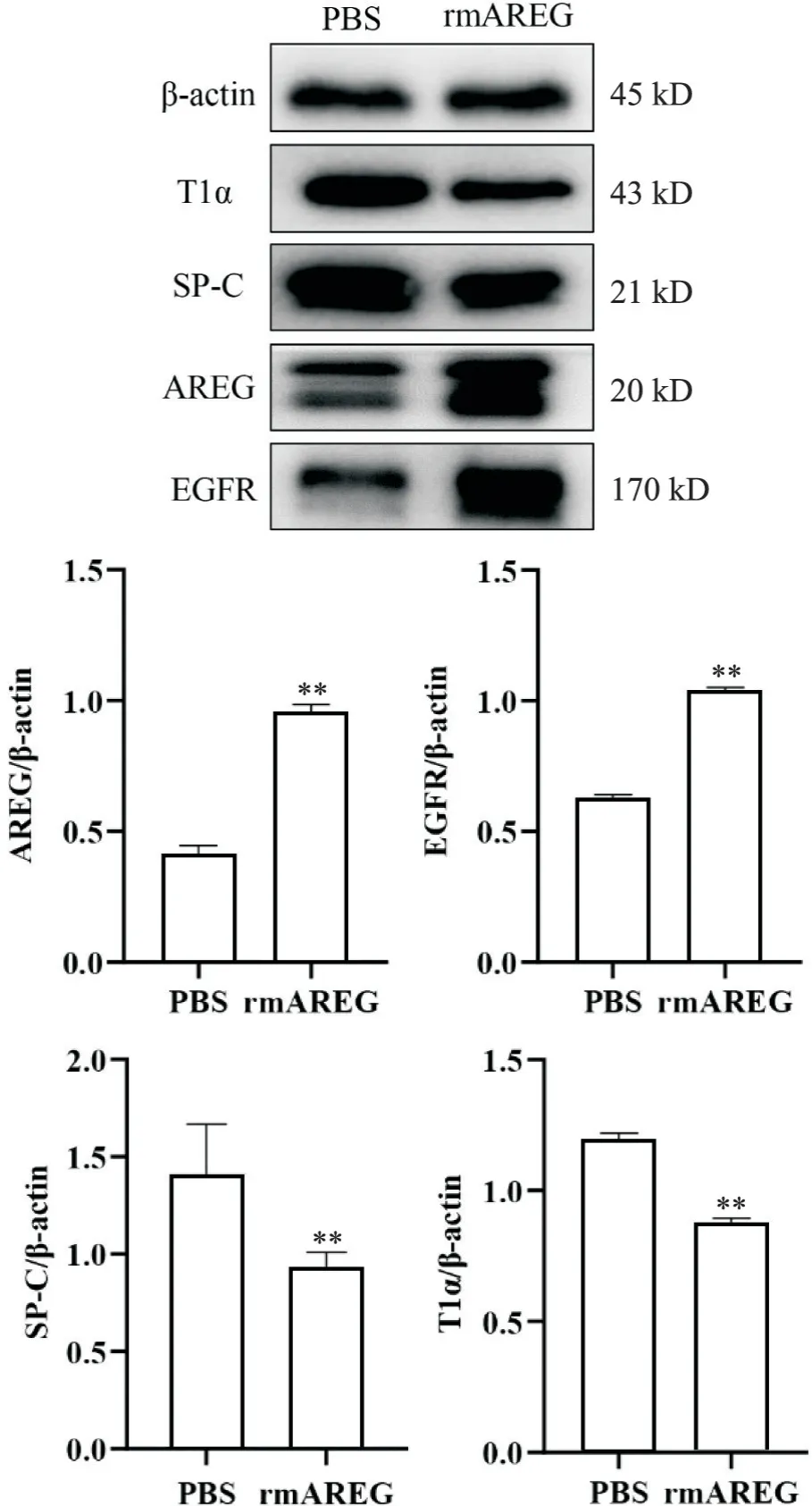
Figure 3. The protein expression of AREG, EGFR, SP-C and T1α in mice lung tissues after overexpression of AREG under hyperoxia. Mean±SD. n=5. **P<0.01 vs PBS group.图3 高氧下过表达AREG后小鼠肺组织AREG、EGFR、SP-C及T1α的蛋白表达水平
6 高氧暴露BPD小鼠敲低AREG后肺组织中SP-C和T1α免疫荧光共定位情况
在高氧状态下,小鼠鼻内滴注AREG siRNA慢病毒表达载体溶液后,与NC组相比,KD组中SP-C与T1α共定位细胞显著增多(P<0.01),见图6。
讨 论
BPD是早产儿最常见的并发症之一,其主要病理学特征是肺泡化受阻,表现为AECII向AECI分化障碍[10]。AREG在免疫调节中有重要作用,是参与炎症细胞中肌成纤维细胞分化的驱动因素[15]。本研究通过建立高氧暴露的BPD模型小鼠,过表达及敲低AREG并观察其对肺泡分化的影响,结果显示AREG抑制高氧暴露的BPD模型小鼠AECII向AECI分化。
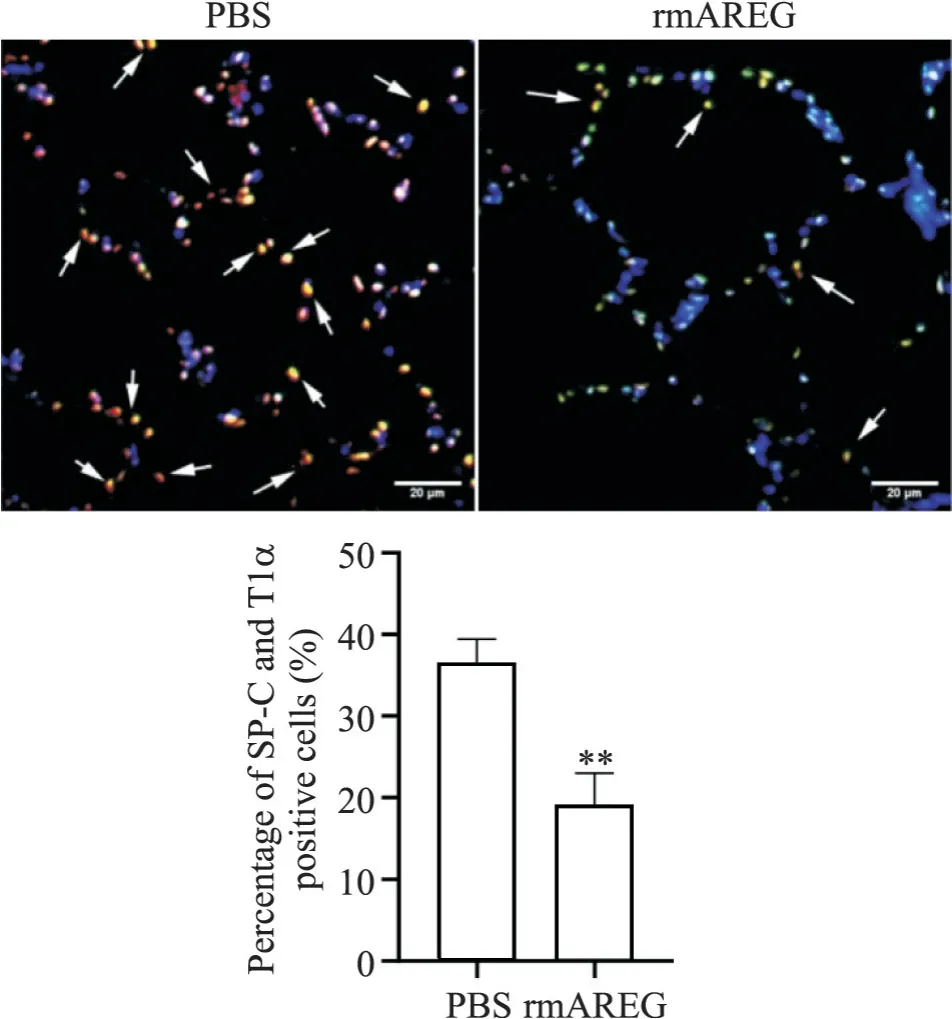
Figure 4. Immunofluorescence staining of SP-C (green) and T1α (red) in mice lung tissues after overexpression of AREG under hyperoxia(scale bar=20 μm). Blue indicates DAPI; arrows indicate SP-C and T1α positive cells. Mean±SD. n=5. **P<0.01 vs PBS group.图4 高氧下过表达AREG后小鼠肺组织中SP-C和T1α免疫荧光共定位细胞情况
肺发育是一个复杂的过程,可分为胚胎期、假腺体期、小管期、囊泡期和肺泡期[16]。在小管期和囊泡期阶段,肺细胞开始分化,末端分支变窄并形成上皮囊,在出生后肺泡阶段发育成肺泡[17]。BPD的主要病理变化发生在囊泡期和肺泡期,肺发育中断,肺泡化过程受阻[18-19]。本研究通过对高氧暴露BPD模型小鼠肺上皮标志物研究,观察到与同时点空气组相比,高氧组SP-C蛋白表达降低,T1α蛋白表达增加,提示高氧后AECII减少而AECI增加,但免疫荧光双标显示SP-C与T1α共定位细胞并未增加,反而减少,提示高氧下AECII向AECI分化减少。由于实验条件所限,本研究未能对AECII进行示踪研究,但有研究通过对AECII进行特殊标记,观察到在高氧状态下,T1α的增多并不是由已标记的AECII分化而来[20],且有研究显示,在高氧状态下,尽管T1α蛋白表达增加,但在损伤修复时AECI功能受损,这可能与肺上皮紧密连接破坏以及气血屏障受损有关[21],结合本次研究结果,提示在高氧状态下存在AECII向AECI分化障碍。
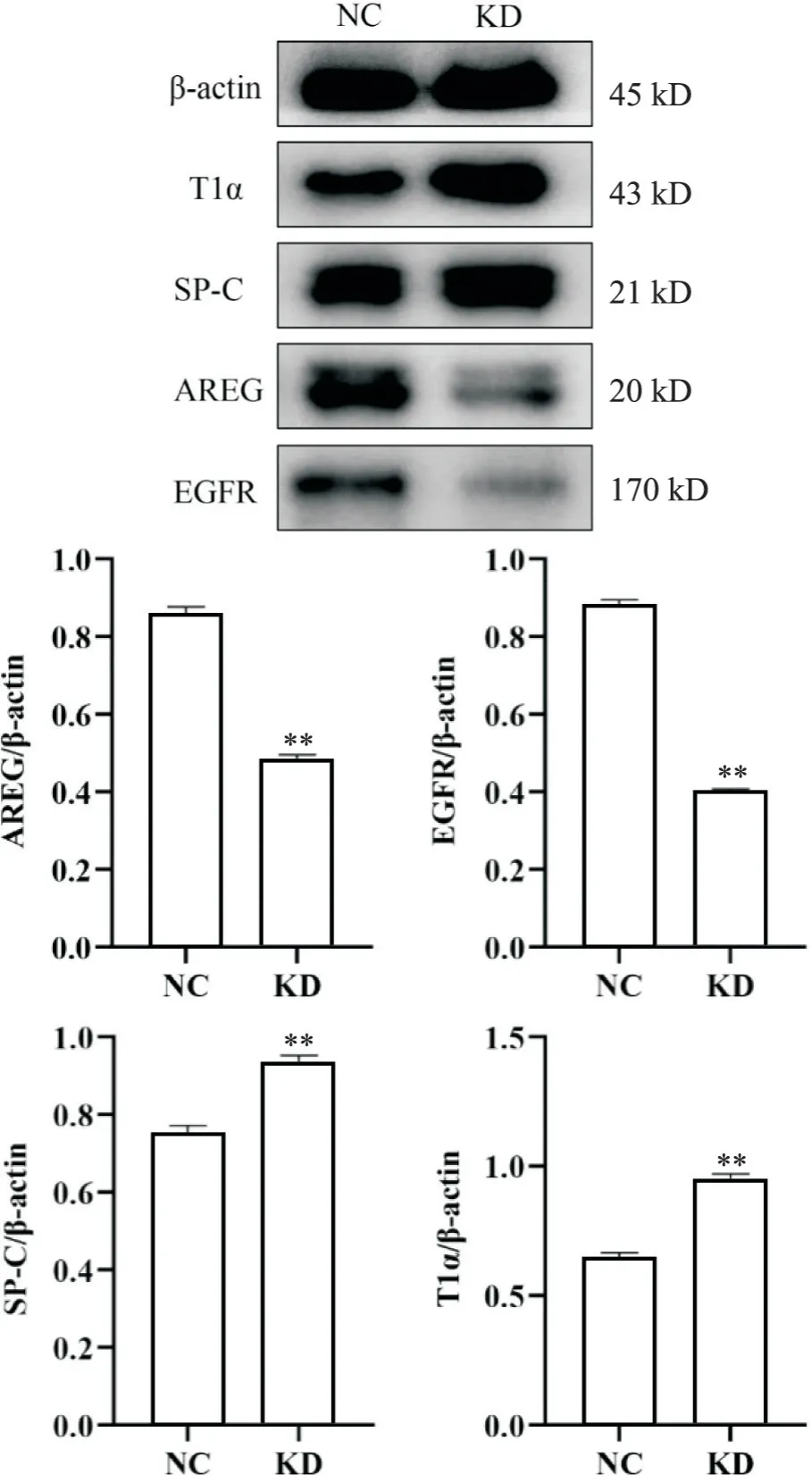
Figure 5. The protein expression of AREG, EGFR, SP-C and T1α in mice lung tissues after knockdown of AREG under hyperoxia. Mean±SD. n=5. **P<0.01 vs NC group.图5 高氧下敲低AREG后小鼠肺组织中AREG、EGFR、SPC及T1α的蛋白表达水平
AREG在上皮细胞生长和分化中作用的研究引起关注。Hillman等[22]研究显示,在早产羔羊机械通气期间,肺组织AREG表达随着机械通气时间增多而升高,并影响肺泡分化。本研究观察到高氧组中AREG表达较同时点空气组升高,提示AREG参与了BPD的病理生理过程。Zuo等[23]研究显示,AREG可驱动气道上皮细胞向基质细胞和黏膜细胞增生分化,但AREG是否参与BPD肺泡分化障碍尚不明确。
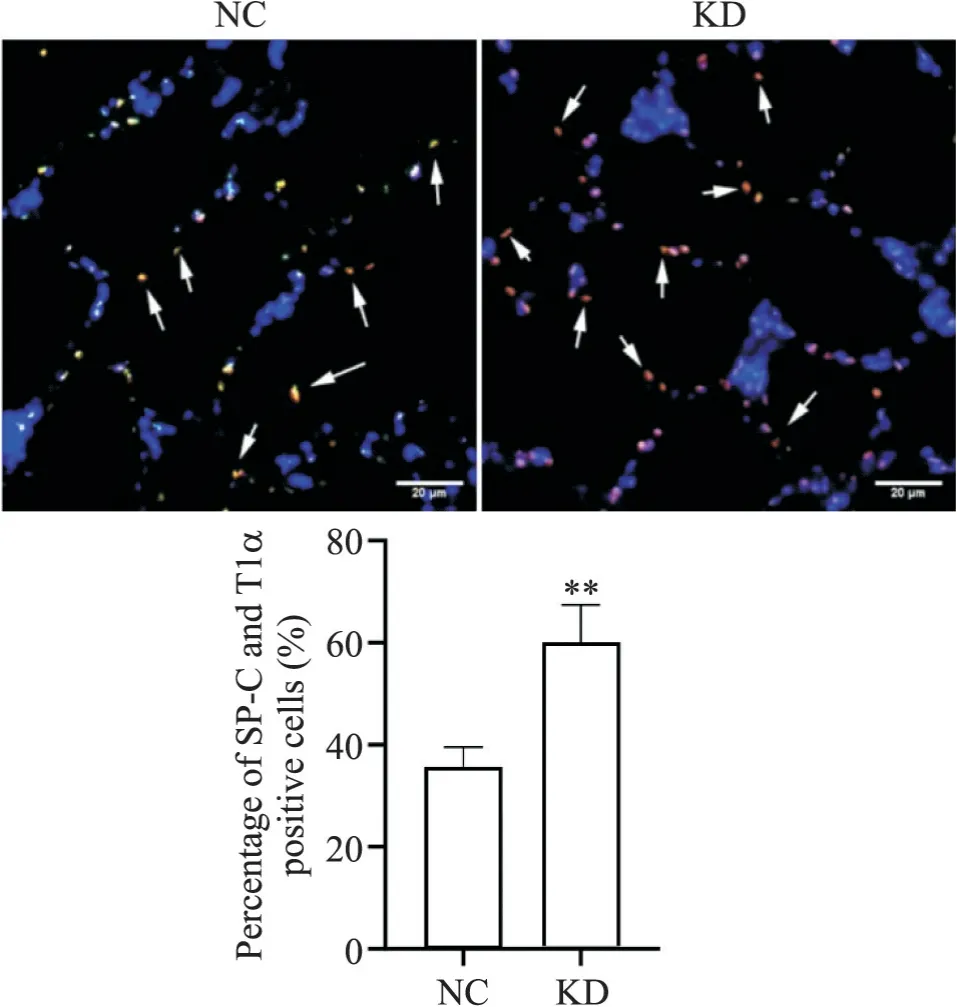
Figure 6. Immunofluorescence staining of SP-C (green) and T1α (red) in mice lung tissues after knockdown of AREG under hyperoxia(scale bar=20 μm). Blue indicates DAPI; arrows indicate SP-C and T1α positive cells. Mean±SD. n=5. **P<0.01 vs NC group.图6 高氧下敲低AREG后小鼠肺组织SP-C和T1α的免疫荧光共定位情况
为进一步探讨AREG表达变化对高氧暴露BPD模型小鼠肺泡分化的影响,本研究通过腹腔注射rmAREG蛋白实现AREG过表达,鼻内滴注AREG siRNA慢病毒表达载体溶液实验AREG敲低,并检测肺组织中SP-C和T1α表达变化,观察到过表达AREG后,SP-C蛋白及T1α蛋白表达减少,SP-C与T1α共定位细胞数显著减少,表明BPD小鼠过表达AREG后AECII向AECI分化障碍加重。在肺慢性异体移植功能障碍的气道重塑中,观察到AREG主要定位于气道上皮细胞,且过表达AREG后,出现纤毛细胞分化,纤维化增加[24]。而在西地那非治疗的BPD模型大鼠中,AREG mRNA的表达降低,肺泡化障碍得以改善[25]。本研究显示,敲低AREG后,SP-C和T1α蛋白表达增加,SP-C与T1α共定位细胞增多,也进一步证实AREG减少可缓解高氧暴露BPD小鼠中AECII向AECI分化障碍。在转化生长因子β诱导的肺纤维化发病机制中,AREG的沉默会降低成纤维细胞的增殖、肌成纤细胞的转化及胶原沉积[26],在萘诱导的肺损伤模型中也显示AREG是肺上皮黏液细胞化生的关键因子,AREG敲除后肺上皮黏液细胞化生得到缓解[27],这与本研究一致,提示AREG可通过影响AECII向AECI分化参与BPD的发展。在病理状态下,肺上皮细胞可通过上皮-间质转化为基质成纤维细胞或肌成纤维细胞,导致BPD的异常修复[28],但AREG是否参与BPD上皮-间质转化仍有待进一步研究。
Lkhagvadorj等[10]研究表明,AREG/EGFR信号通路与受损的上皮细胞分化有关,作为AREG的受体,EGFR可调节肺损伤后细胞的生长、分化、迁移以及细胞间黏附[29]。本研究显示高氧组EGFR表达升高,且随着高氧时间的延长,EGFR表达呈上升趋势,提示在高氧状态下,EGFR增多以响应AREG/EGFR信号转导。本实验进一步研究观察到过表达AREG后,EGFR蛋白表达升高,而敲低AREG后,EGFR蛋白表达降低,提示EGFR表达趋势与AREG一致。Todd等[24]研究显示,在肺移植后的气道重塑中,AREG和EGFR转录组在组织中大量表达,且定位于富有纤维组织的气道上皮细胞,也有研究观察到脂多糖(lipopolysaccharide,LPS)诱导的BPD大鼠模型中,EGFR的增加影响了肺肌成纤维细胞的定位,导致BPD中肺泡发育受阻[30]。综合以上研究,推测AECII向AECI分化障碍与AREG/EGFR信号通路有关,该机制可能在BPD模型小鼠肺泡分化障碍机制中起重要作用,但仍需进一步研究。
综上所述,本研究初步探讨了AREG对BPD模型小鼠肺泡分化的影响,观察到高氧暴露BPD模型小鼠肺组织AREG升高,AECII向AECI分化障碍;高氧下敲低AREG后,AECII向AECI分化障碍缓解,提示AREG参与了BPD模型小鼠肺泡分化障碍的病理生理过程,可能与AREG抑制AECII向AECI分化有关。本研究的局限性在于未能结合AECII示踪技术探讨肺上皮细胞分化,且仅通过腹腔注射外源性重组AREG蛋白及AREG慢病毒实现AREG过表达及敲低,未能通过基因敲除技术敲除小鼠AREG进行研究,这些将在今后的研究中进一步优化。
猜你喜欢
广东医学(2022年5期)2022-06-01
清华金融评论(2022年4期)2022-04-13
食品安全导刊(2022年2期)2022-03-18
天津医科大学学报(2021年4期)2021-08-21
现代临床医学(2021年4期)2021-07-31
国际放射医学核医学杂志(2021年10期)2021-02-28
房地产导刊(2020年7期)2020-08-24
国际呼吸杂志(2019年8期)2019-04-29
国际呼吸杂志(2019年8期)2019-04-29
中国医药导报(2018年36期)2018-03-04
