
核壳结构Ni@SiO2 催化剂的制备及其在甲烷二氧化碳重整反应中的应用
2023-01-31 04:48贺露露任远航贺鹤勇
复旦学报(自然科学版) 2022年6期
贺露露,陈 欣,任远航,岳 斌,贺鹤勇
(复旦大学 化学系 上海市分子催化和功能材料重点实验室,上海 200438)
甲烷二氧化碳重整反应不仅可以改变现有的以煤、石油为主的能源结构,增加清洁能源的使用占比,还可以减少温室气体排放,且该反应的主要产物合成气的H2/CO 的比例接近于1,后续更利于通过Fischer-Tropsch反应转化为高附加值的液体燃料和化学品[1-2]。贵金属基催化剂在甲烷二氧化碳重整反应中具有优异的催化性能,但有限的资源和高的成本使其难以在工业中广泛应用[3-5]。因此,人们对非贵金属催化剂,特别是镍(Ni)基催化剂开展了广泛的研究[6-8]。相比贵金属催化剂,镍基催化剂需要解决严重的积碳和烧结问题[9-11]。除了镍基组分本身,载体的性质对Ni基催化剂的抗积碳性能十分重要[12-14]。大比表面积的载体利于活性金属的分散和催化剂稳定性的提高[9]。载体和活性金属间的相互作用、载体表面的氧化还原性等亦影响抗积碳和抗烧结性能[15]。近年来,二氧化硅(SiO2)载体因其具有高比表面积和可调的孔径而受到广泛的关注[16-17]。在高度有序多孔二氧化硅载体的研究基础上,人们发现核壳结构SiO2封装Ni的催化剂可大大降低金属Ni的烧结和阻止积碳的形成[18-22],但积碳和烧结问题仍未得到完全解决,催化剂的性能还需进一步提高。文献报道,碱处理可改变二氧化硅材料的结构,改善其催化性能。Zhang等[23]发现,用KOH 处理负载Ni的凹凸棒石,可提高催化剂的比表面积和微孔量,活化过程中可增加氧化镍的还原度和镍物种的分散性,使得活化后的催化剂在乙酸和愈创木酚蒸汽重整反应中具有优异的催化活性。Chen等[24]报道Ni/SBA-15通过氨水热处理和高温处理可调变催化剂的织构性质和金属-载体相互作用,从而改变催化剂在甲烷二氧化碳重整反应中的催化及抗积碳性能。
本文中,合成的核壳结构的Ni@SiO2通过正丁胺碱蒸汽处理后在高温焙烧得到重构的催化剂,采用多种表征技术研究了碱蒸汽处理对催化剂化学性质的影响,重构后的催化剂在甲烷二氧化碳重整反应中表现出优异的催化性能、抗金属烧结和抗积碳性能。
1 实验部分
1.1 Ni@SiO2 的制备
含NiO 纳米粒子的核壳结构Ni@SiO2采用一锅反相胶束法制备。室温下将20.16 g聚乙二醇单-4-壬基苯基醚(NP-5,≥98%,TCI试剂有限公司)溶解于480 mL环己烷中。搅拌状态下加入0.27 mol·L-1的Ni(NO3)2水溶液2.16 mL。35℃下搅拌15 h后快速加入2.16 mL浓氨水,继续搅拌2 h。用微量进样泵缓慢加入2.5 mL正硅酸乙酯(TEOS,分析纯,CROSS试剂公司)到上述溶液中,继续搅拌48 h。反应结束后,以乙醇为去稳定剂并在10 000 rpm 下离心收集催化剂前驱体。将收集到的样品放入40℃的真空烘箱中干燥12 h,最后置于马弗炉中在800℃焙烧4 h得到Ni含量为5.0%(质量分数)的Ni@SiO2。
1.2 Ni@SiO2-B的制备
碱蒸汽处理在150 mL带有聚四氟乙烯内衬的反应釜中进行。反应釜内放入30 mL 20%的正丁胺溶液,真空干燥且未焙烧的前驱体Ni@SiO2放入一带孔的聚四氟乙烯容器并置于反应釜中,正丁胺溶液与固体样品无接触。200℃下水热反应120 h,正丁胺蒸汽与固体样品充分接触反应后冷却至室温,取出固体样品用去离子水洗至中性,真空烘箱40 ℃干燥12 h,然后在马弗炉中在800 ℃焙烧4 h,样品记为Ni@SiO2-B。除注明外,以上试剂均为分析纯,来自国药集团化学试剂有限公司。
1.3 催化剂表征
X射线衍射(X-Ray Diffraction,XRD)分析采用Bruker D8 Advance衍射仪,Cu Kα射线(λ=1.541 8Å),工作电压为40 kV,工作电流为40 mA,采集的范围10°<2θ<80°,步长0.02°,停留时间0.6 s。体相组成的测定使用Thermo Elemental IRIS Intrepid 电感耦合等离子体原子发射光谱仪(Inductive Coupled Plasma-Atomic Emission Spectrometer,ICP-AES)。比表面积、孔体积和平均孔径测定在Micromeritics Tristar 3000吸附仪上进行,样品首先在真空状态下250℃脱气3 h,然后在-196℃下测定N2吸附-脱附等温线,采用Brunauer-Emmett-Teller(BET)方法计算比表面积,通过脱附分支和Barrett-Joyner-Halenda(BJH)模型计算孔体积和平均孔径。透射电子显微镜(Transmission Electron Microscope,TEM)、高分辨透射电子显微镜(High Resdution Transmission Electron Microscope,HRTEM)和能量色散X 射线光谱(Energy Dispersive X-ray Spectroscopy,EDX)分别在JOEL JEM-2011透射电子显微镜、FEI Tecnai G2 F20 S-TWIN 型高分辨场发射透射电子显微镜及其后者自带的能量散射X 射线分析仪测定。傅里叶变换红外光谱(FT-IR)采用Thermo Fisher Nicolet iS10光谱仪,KBr粉末混合压片,分辨率4 cm-1。热重-差热分析(TG/DTA)采用TA SDT Q600型集成热分析仪,空气流速为100 mL min-1下以10℃min-1加热 至900 ℃。氢 气-程序升温脱附和还原(H2-TPD 和H2-TPR)均在Micromeritics ChemiSorb 2720化学吸附仪上进行。H2-TPD 测试时,样品先在一定温度下用10% H2/Ar混合气(50 mL min-1)还原90 min,Ar气氛下冷却至40℃后,样品在40℃吸附H20.5 h,再用Ar吹扫0.5 h 去除表面物理吸附H2,10℃min-1升至750℃,Ag2O 为标样计算H2-TPD 的脱附量,假定吸附氢原子与表面金属镍原子的比为1∶1计算金属Ni的分散度。H2-TPR 测试时,样品先用Ar(50 mL min-1)吹扫40 min,再在200℃下Ar中脱气2 h,冷却至室温后换成10% H2/Ar,测定范围为室温至900℃。
1.4 甲烷二氧化碳重整反应催化性能评价
将30 mg 40~60目催化剂与相同目数的石英砂混合均匀后置于内径为5 mm 的固定床石英管反应器中,800℃H2还原90 min后,通入Ar吹扫并降至所需实验温度。反应空速(GHSV)为24 000 mL h-1g-1,CH4∶CO2∶Ar的体积比为1∶1∶3,常压,在线色谱配热导检测器分析。
2 结果与讨论
2.1 催化剂表征
Ni@SiO2和Ni@SiO2-B 的氮吸附-脱附等温线(见图1)均具为Ⅳ型等温线[25]及H3型滞后环[26],表明存在较为规则的介孔。Ni@SiO2的BET比表面积、孔体积和平均孔径分别为128 m2·g-1、0.52 cm3·g-1和11.7 nm,Ni@SiO2-B的BET 比表面积、孔体积和平均孔径分别为123 m2·g-1、0.50 cm3·g-1和13.9 nm,Ni@SiO2-B可能由于碱蒸汽处理引起表面重构而导致比表面积和孔体积略有减少、平均孔径增大[24]。ICP-AES测得的Ni@SiO2和Ni@SiO2-B的实际Ni含量分别为4.7%和4.8%(质量分数),和理论值基本一致。
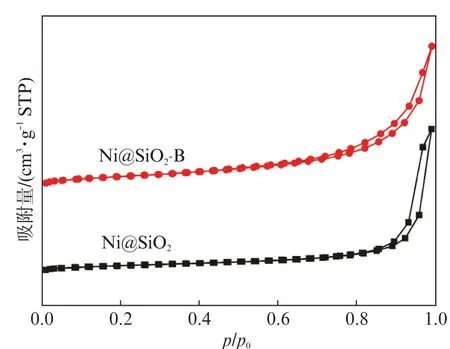
图1 Ni@SiO2 和Ni@SiO2-B的N2 吸附-脱附等温线Fig.1 N2 adsorption-desorption isotherms of the Ni@SiO2 and Ni@SiO2-B samples
图2(a)显示Ni@SiO2和Ni@SiO2-B均在2θ约为22°处具有一个较宽的衍射峰,归属于无定型二氧化硅[27]。Ni@SiO2还在2θ为37.3°、43.3°和63.0°出现衍射峰,分别可归属于立方NiO 相(PDF No.04-0835)的(111)、(200)和(220)面的衍射,即Ni@SiO2中Ni主要为NiO。利用NiO 的(200)衍射峰的半峰宽和Scherrer公式,可估算NiO 的颗粒尺寸约为3.0 nm。而Ni@SiO2-B未观察到NiO 衍射峰,但在2θ为34.1°、36.7°和60.9°处出现了归属于蛇纹石型硅酸镍Ni3(Si2O5)(OH)4(PDF No.49-1859)的(200)、(202)和(060)面的衍射峰[28-29],说明Ni@SiO2在碱蒸汽处理中发生了重构现象,在高温焙烧后形成的Ni@SiO2-B中Ni物种主要存在于硅酸镍相中。
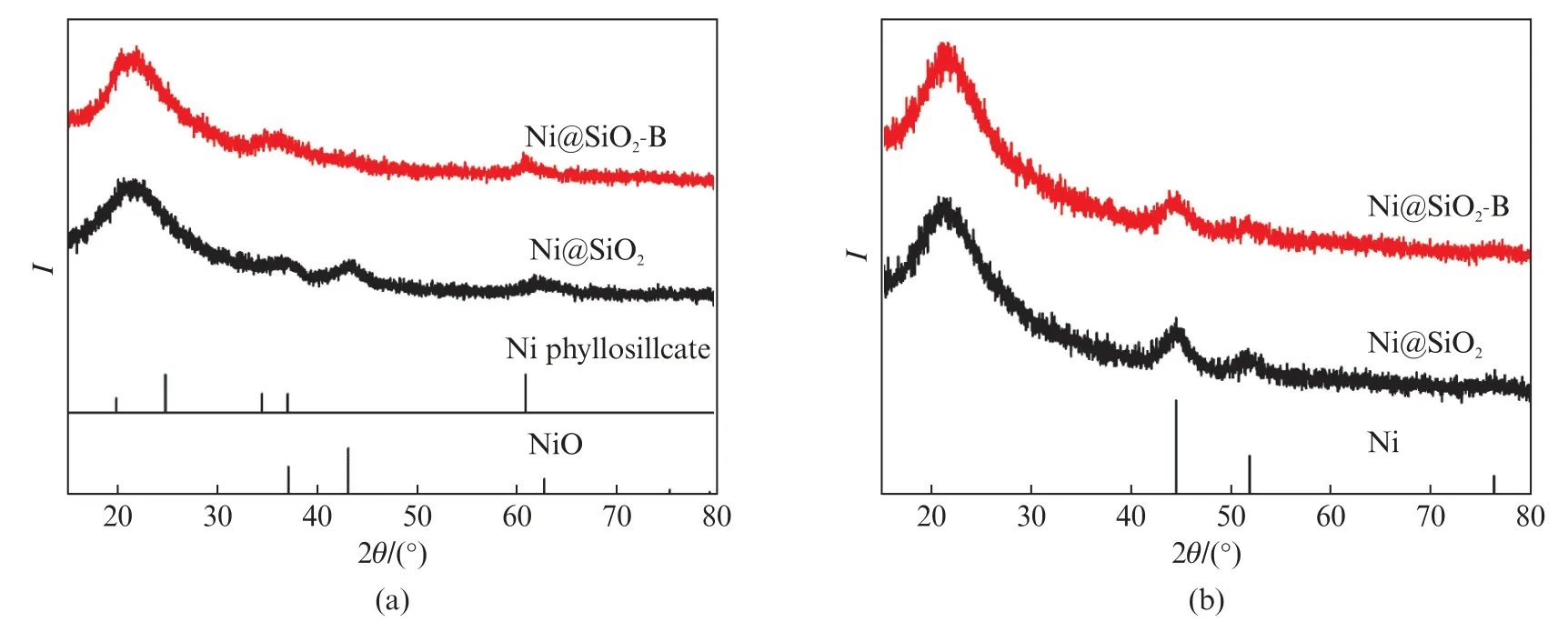
图2 (a)Ni@SiO2 和Ni@SiO2-B及(b)其H2 还原后的XRD 图谱Fig.2 XRD patterns of(a)the Ni@SiO2 and Ni@SiO2-B samples and(b)their counterparts after reduction in H2
图3(a)给出了Ni@SiO2的TEM 图和NiO的粒径分布图。从TEM 图可见,由反相胶束法合成的SiO2壳基本呈现出单分散的球状,二氧化硅球体表面相对光滑,平均直径约为30 nm。NiO 颗粒被均匀地包裹在SiO2球体中,呈高度分散状态。对200个NiO 颗粒进行粒径统计,可得出其平均粒径约为3.2 nm,与通过XRD图谱计算出来的NiO粒径基本一致。图3(b)给出了Ni@SiO2-B的TEM 图,经碱蒸汽处理后的样品发生了一定的重构,SiO2壳的球形基本保持,但在球的表面形成了一些针状的细枝,此特征微结构也证实了硅酸镍的存在。由于无明显NiO颗粒,无法获取Ni@SiO2-B中镍组分的粒径。文献报道,NiO 参与形成硅酸镍后,Ni与SiO2间的相互作用增强,导致形成的镍颗粒过小[30],所以难以观察到NiO的存在。

图3 (a)Ni@SiO2 的TEM 图以及NiO 粒径分布图,(b)Ni@SiO2-B的TEM 图Fig.3 (a)TEM image and NiO particle size distribution of the Ni@SiO2,(b)TEM image of the Ni@SiO2-B
当高能电子束直接照射到被测样品时,高能电子可还原硅酸镍形成镍纳米颗粒[28]。图4(见第702页)分别是Ni@SiO2-B暴露在200 kV 高能电子束5 min和7 min所测得的HRTEM 图,可以看到,“纤维状”的硅酸镍转化为Ni纳米颗粒的暗点,且还原后形成的Ni纳米颗粒粒径较小,且呈现出高度分散的状态。EDS的面扫分析可知,Ni@SiO2中Ni物种较均匀分布在SiO2上,但在高能电子束照射下,Ni纳米颗粒发生一定的团聚。Ni@SiO2-B中Ni物种高度分散,说明硅酸镍的形成增强了Ni和SiO2间的金属-载体相互作用,使得Ni@SiO2-B中活性物种分散度高且稳定。

图4 (a)5分钟和(b)7分钟测试后Ni@SiO2-B在的HRTEM 图Fig.4 HRTEM images of the Ni@SiO2-B under electron beam for(a)5 min and(b)7 min
Ni@SiO2和Ni@SiO2-B在H2气氛下800 ℃还原90 min后的XRD 图谱(图2(b))可见,归属于NiO和硅酸镍的衍射峰消失,在2θ为44.5°和51.8°处出现了两个新的衍射峰,分别可归属于金属Ni(PDF No.04-0850)的(111)和(200)晶面的衍射[30-31]。表1中列出了采用Scherrer公式及Ni(111)半峰宽估算的还原后样品的Ni平均粒径,可见还原后Ni@SiO2和Ni@SiO2-B的Ni平均粒径分别为4.2和3.9 nm。表1中通过H2-TPD测得的还原后样品Ni的分散度结果显示,Ni@SiO2-B的Ni分散度略高于Ni@SiO2。还原后样品的XRD和H2-TPD结果表明,碱蒸汽处理的Ni@SiO2-B具有更高的镍分散度且镍颗粒更小,这可能是因为经碱蒸汽处理后的Ni@SiO2-B生成了硅酸镍,增强了活性金属与载体间的相互作用。具有较小Ni纳米颗粒的催化剂有利于提高甲烷二氧化碳重整反应的催化性能和抗积碳性能。
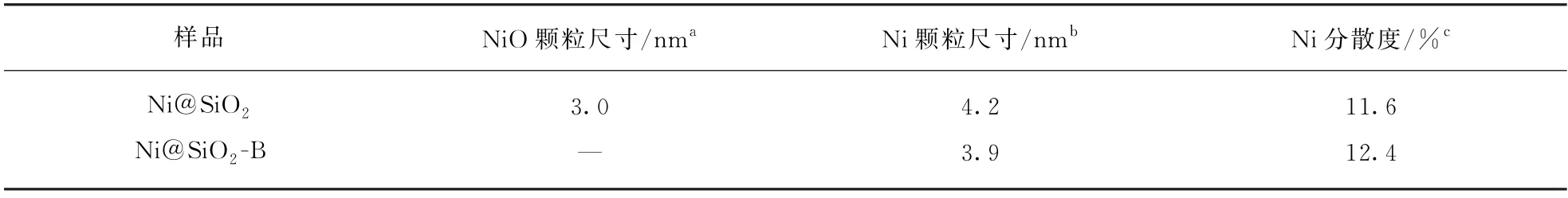
表1 Ni@SiO2 和Ni@SiO2-B的颗粒尺寸及分散度Tab.1 Crystallite size and Ni dispersion for the Ni@SiO2 and Ni@SiO2-B samples
样品的氧化还原性及Ni与SiO2载体间的相互作用采用H2-TPR 技术表征。如图5所示,Ni@SiO2具有3个还原峰,位于350~470℃区间的还原峰可归属为NiO 颗粒的还原[32],470~600℃间的还原峰来自与载体相互作用较弱的Ni2+的还原,600~750℃还原峰可归属于与SiO2载体相互作用较强的Ni2+的还原,最后这部分的Ni物种与SiO2产生较强的金属-载体相互作用[29]。文献表明,在600~750℃高温区的还原峰可能是Ni与SiO2载体形成了硅酸镍物种,但Ni@SiO2的XRD 图谱中并未发现归属于层状硅酸镍的衍射峰,在Ni@SiO2中Ni物种应该主要还是以具有不同金属-载体相互作用的NiO 的形式存在[33-34]。而对于Ni@SiO2-B,可以明显看出,样品处于中低温区间的还原峰面积减少。绝大多数Ni物种的还原都在高温区。XRD 结果证明,碱蒸汽及高温焙烧处理后Ni@SiO22-B 中Ni主要是以Ni3(Si2O5)(OH)4存在[20,32],这种结构的形成增强了Ni与载体间的相互作用,导致还原温度偏向高温区域,同时较强的金属-载体间相互作用可促进Ni的分散,抑制催化剂中Ni颗粒的团聚与烧结,从而保持更小的金属Ni纳米颗粒尺寸,有利于甲烷二氧化碳重整反应的进行。

图5 Ni@SiO2 和Ni@SiO2-B的H2-TPR 结果Fig.5 H2-TPR profiles of the Ni@SiO2 and Ni@SiO2-B samples
图6给出了样品的FT-IR 图谱。两种样品均在810和1 100 cm-1处出现了两个峰,分别归属于硅氧四面体结构单元中Si—O—Si键的弯曲振动和伸缩振动峰,1 630和3 640 cm-1处两个峰分别对应于吸附水的H—O—H 的伸缩振动和弯曲振动峰[29,35]。Ni@SiO2在970 cm-1附近处出现了归属于表面Si—O—H 伸缩振动峰[36]。Ni@SiO2-B在670、710和3625cm-1附件处出现了3个新的峰,其中位于670和710cm-1处的峰分别对应于Ni3(Si2O5)(OH)4中的四面体Si—O振动和连接到Ni原子上孤立OH 基团的弯曲振动[37-38]。而在3625cm-1的峰是由Ni3(Si2O5)(OH)4中OH 基团的伸缩振动引起的[39]。此外,Ni@SiO2-B 的谱图上未观察到位于1 024和3 645cm-1处归属于Ni3(Si2O5)2(OH)2的硅酸镍谱带[37],证明样品中Ni物种主要是以Ni3(Si2O5)(OH)4的形式存在。
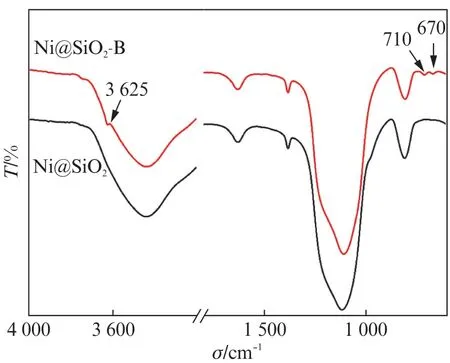
图6 Ni@SiO2 和Ni@SiO2-B的红外光谱图Fig.6 FT-IR spectra of the Ni@SiO2 and Ni@SiO2-B samples
2.2 甲烷二氧化碳重整反应催化性能
2.2.1 催化剂在不同反应温度下的反应活性
首先在550~800℃范围内评价Ni@SiO2和Ni@SiO2-B催化剂的甲烷二氧化碳重整反应活性。如图7所示,由于甲烷二氧化碳重整反应是强吸热反应,因此,CH4和CO2的转化率随反应温度升高而升高。而且,由于同时发生了逆水煤气变换反应(CO2+H2→CO+H2O),所以催化剂的CO2转化率略高于CH4转化率[7,14,21]。在不同的反应温度下,Ni@SiO2-B 催化剂的CH4和CO2的转化率均略高于Ni@SiO2,在800℃下CH4和CO2的最高转化率分别可达95.1%和97.3%。这显示经过碱蒸汽处理再高温焙烧后的Ni@SiO2-B催化剂形成了更强的金属-载体相互作用,使得催化剂上的活性Ni物种分散度更高,在还原过程中能得到较小的Ni颗粒,表现出了更高的催化性能。
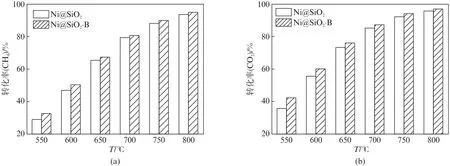
图7 不同反应温度下Ni@SiO2 和Ni@SiO2-B催化甲烷二氧化碳重整反应的(a)CH4 转化率和(b)CO2 转化率Fig.7 (a)CH4 and(b)CO2 conversions of the Ni@SiO2 and Ni@SiO2-B catalysts at various reaction temperatures in methane dry reforming reaction
2.2.2 催化剂稳定性研究
图8给出了Ni@SiO2和Ni@SiO2-B催化剂在700℃、0.1 MPa和GHSV 为24 000 mL g-1h-1条件下100 h的甲烷二氧化碳重整反应的催化稳定性。Ni@SiO2-B催化剂初始反应活性略高于Ni@SiO2,与图7结果一致。随着反应的进行,Ni@SiO2催化剂的活性发生了明显的下降,CH4转化率从79.5%降为71.8%,CO2转化率从85.9%降为80.1%。而Ni@SiO2-B催化剂表现出优异的催化稳定性,在100 h的反应过程中观察不到CH4和CO2转化率的明显下降。
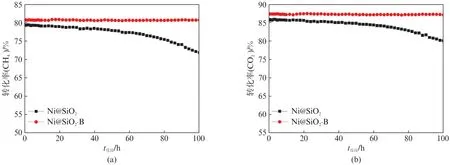
图8 Ni@SiO2和Ni@SiO2-B催化甲烷二氧化碳重整反应100 h的(a)CH4 转化率和(b)CO2 转化率稳定性结果Fig.8 (a)CH4 and(b)CO2 conversions of the Ni@SiO2 and Ni@SiO2-B catalysts in 100 h stability tests in methane dry reforming reaction
为了进一步研究Ni@SiO2-B催化剂在不同的反应温度下更长时间的稳定性性能,分别在700℃和650℃下进行了200 h的甲烷二氧化碳重整反应测试。如图9所示,Ni@SiO2-B催化剂在不同的反应温度下均具有优异的催化稳定性,在200 h的反应中几乎观察不到反应活性的下降,700℃和650℃下的CH4和CO2的转化率分别约为80.6%、87.4%和67.7%、76.7%。所得产物的H2/CO 的比例分别约为0.93和0.88,在200 h的测试中也未发现明显的改变。Ni@SiO2-B催化剂因其具有较小的活性金属颗粒和强的金属-载体相互作用,使得催化剂不易金属烧结和产生积碳,从而能保持优异的催化稳定性。
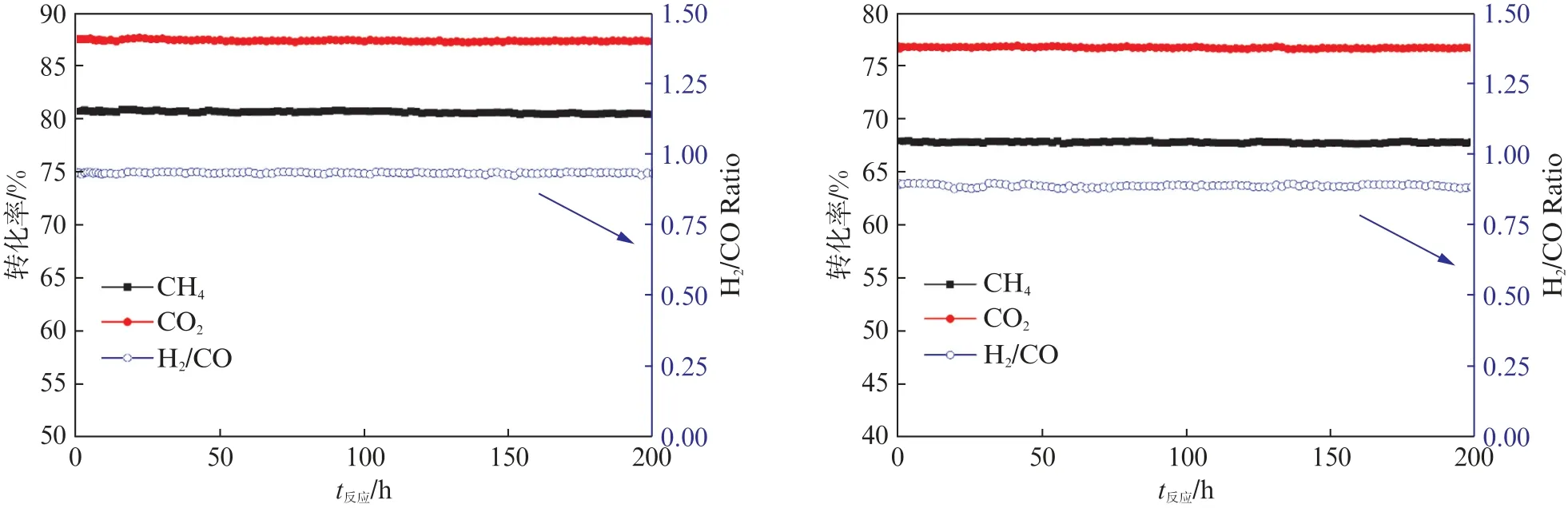
图9 Ni@SiO2-B 催化剂在(a)700℃和(b)650℃下200 h的甲烷二氧化碳重整反应稳定性结果Fig.9 CH4 and CO2 conversions and H2/CO ratio of the Ni@SiO2-B catalyst in 200 h stability test in methane dry reforming reaction at(a)700℃and(b)650℃
2.3 甲烷二氧化碳重整反应后催化剂的表征
为了研究两种催化剂的抗积碳性能,对100 h反应后Ni@SiO2和Ni@SiO2-B 催化剂进行了TG/DTA 分析。图10(a)的TG 结果中,低于200℃的重量变化与催化剂表面吸附水或其他有机物的去除有关,200~600℃之间的重量变化对应于无定形和丝状碳的氧化,高于600℃的重量变化则可归因于石墨碳的氧化[40-41]。从TG 结果计算发现,100 h反应后Ni@SiO2催化剂的重量减少7.1%,而Ni@SiO2-B只减少1.3%,表明Ni@SiO2-B催化剂的抗积碳性能大大优于Ni@SiO2。图10(b)给出了反应后催化剂的DTA 图,可见反应后的催化剂均在650℃附近出现了归属于石墨碳的氧化放热峰。此外,反应后的催化剂还在300和500℃附近出现两个较弱的放热峰,分别对应于无定形碳和丝状碳的氧化[42]。
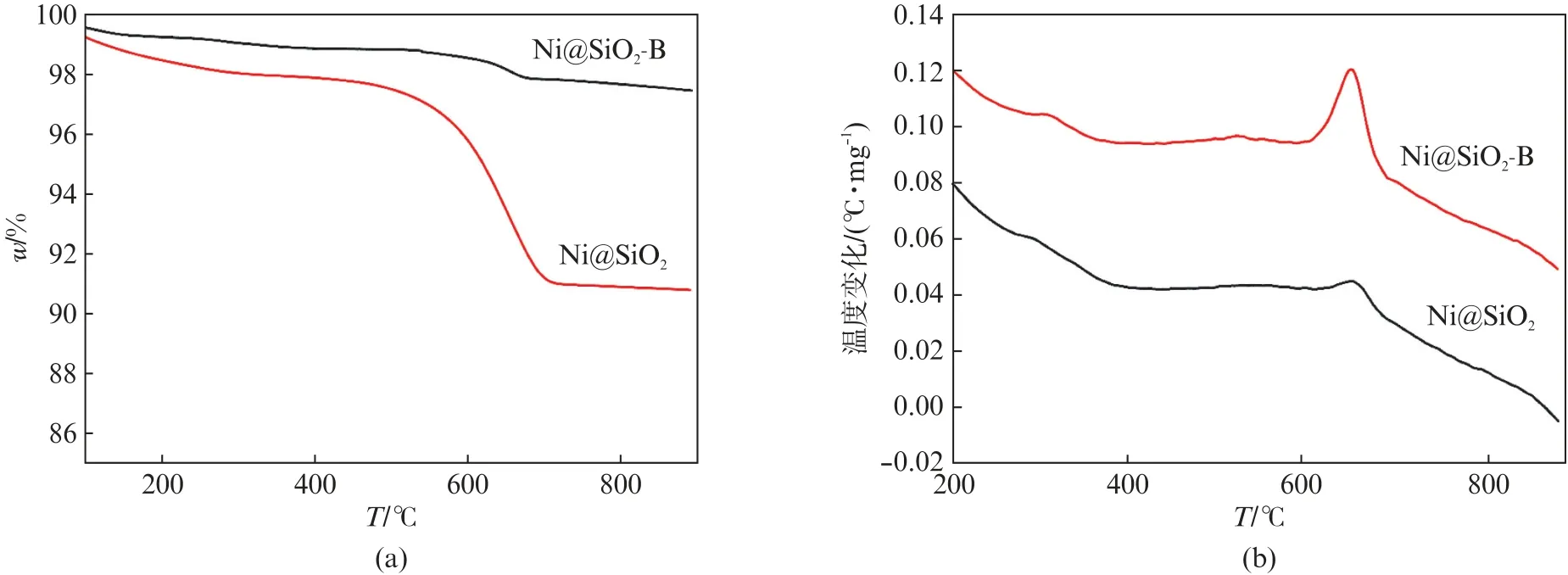
图10 Ni@SiO2 和Ni@SiO2-B催化剂在100 h反应后的(a)TG 图和(b)DTA 图Fig.10 (a)TG and(b)DTA profiles of the Ni@SiO2 and Ni@SiO2-B catalysts after 100 h stability tests
图11 给出了Ni@SiO2-B 催化剂分别在700 ℃和650℃下进行200 h的甲烷二氧化碳重整反应后的XRD图谱,反应后的催化剂除了在2θ约为22.0°处出现了归属于无定型二氧化硅的衍射峰外,还在2θ为44.5°、51.9°和76.4°处出现了分别对应于金属Ni(PDF No.04-0850)的(111)、(200)和(220)面的衍射峰。反应后催化剂的XRD图谱上并未在26.0°处出现归属于石墨碳的衍射峰。使用Scherrer公式根据Ni(200)晶面的衍射峰的半峰宽估算催化剂的Ni颗粒尺寸,可以得到700℃和650 ℃下经过200 h反应的Ni颗粒尺寸分别为4.3和4.2 nm,相比于反应前催化剂的Ni颗粒尺寸(3.9 nm),反应后Ni颗粒尺寸只有微小幅度的增加。Ni@SiO2-B 催化剂优异的抗积碳和抗烧结性能,使得其在多个温度下具有优异的催化甲烷二氧化碳重整反应稳定性。

图11 Ni@SiO2-B催化剂在700℃和650℃下反应200 h后的XRD 图谱Fig.11 XRD patterns of the Ni@SiO2-B catalyst after 200 h reaction at 700℃and 650℃
采用TG/DTA 进一步对200 h反应后Ni@SiO2-B 催化剂的积碳进行了研究。图12(a)(见第706页)显示,在700℃和650℃下反应200 h后,Ni@SiO2-B催化剂的重量损失均很小,证明了催化剂具有优良的抗积碳性能。图12(b)的DTA 图显示,在700℃和650℃反应后的催化剂均在300℃附近出现了一个放热峰,可将其归属于无定形碳的氧化。与石墨碳相比,无定形碳更易进一步被氧化消除。650℃反应后的催化剂还在500~650℃出现了一个放热峰,对应于介于无定形碳和石墨碳之间的碳物种的氧化。700℃和650℃反应后,催化剂均并未在高温区显示归属于石墨碳的放热峰,说明无石墨化程度较高的碳生成,进一步证明了Ni@SiO2-B催化剂优异的抗积碳性能。
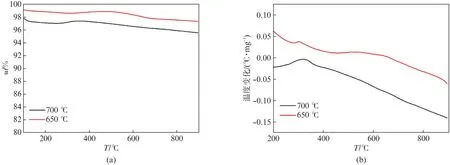
图12 Ni@SiO2-B催化剂在700℃和650℃下反应200 h后的(a)TG 和(b)DTA 图Fig.12 (a)TG and(b)DTA profiles of the Ni@SiO2-B catalyst after 200 h at 700℃and 650℃
3 结论
采用反相胶束法制备了核壳结构Ni@SiO2催化剂,并将核壳结构的催化剂前驱体在20%的正丁胺碱蒸汽中进行处理后在高温下焙烧得到重构后的Ni@SiO2-B催化剂,将两种催化剂用于甲烷二氧化碳重整反应。结果表明,相比于核壳结构Ni@SiO2催化剂,经碱蒸汽处理后Ni@SiO2-B 催化剂发生了重构现象,比表面积和孔体积减小,孔径增大。Ni@SiO2-B 催化剂中活性金属Ni主要以硅酸镍的形式存在,增强了金属-载体相互作用,使得催化剂的还原温度提高,且还原后得到较小的金属Ni纳米颗粒。Ni@SiO2-B催化剂比Ni@SiO2表现出更好的催化活性和稳定性,在700℃和650℃下进行200 h的甲烷二氧化碳重整反应后,Ni@SiO2-B催化剂未有失活现象发生,Ni颗粒粒径无明显增大,且催化剂几乎没有石墨碳生成,表明了该催化剂具有优异的抗金属烧结和抗积碳性能。
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
银行家(2022年5期)2022-05-24
长白学刊(2022年2期)2022-02-05
法制博览(2021年14期)2021-11-25
陶瓷学报(2021年3期)2021-07-22
陶瓷学报(2019年6期)2019-10-27
中国调味品(2017年2期)2017-03-20
中国塑料(2016年10期)2016-06-27
安徽工业大学学报(自然科学版)(2015年4期)2015-12-14
中学化学(2015年2期)2015-06-05
