
灰黄霉素固体分散体在比格犬上的药物代谢动力学分析
2023-01-31 07:30王灵灵王令令张雨晴
中国兽医学报 2022年11期
王灵灵,王令令,张雨晴,马 宁,何 欣
(河北农业大学 动物医学院,河北 保定 071001)
灰黄霉素 (griseofulvin,Gri) 是从灰黄青霉 (penicillium griseofulvin) 培养液中得到的一种含氯代谢产物,是一种非多烯类的抗真菌抗生素[1],为白色或类白色结晶性粉末,无臭,味微苦,在临床上是治疗皮肤真菌病最常用的抗菌药物之一[2]。灰黄霉素在水中极微溶解,仅为13.06 mg/L,在生物药剂学分类系统 (biopharmaceutics classification system,BCS) 中属于低溶解性高渗透性的Ⅱ类药物[3]。由于极低的水溶性,导致需要更大剂量才能达到治疗水平,因此需要改善其水溶性提高抗真菌活性[4]。
固体分散体是指由一种或多种活性成分高度分散在适宜的载体中而形成的一种固体分散体系[5],因颗粒尺寸的减小、团聚的减少和润湿性的改善可显著提高药物的溶解度和生物利用度[6]。常用的固体分散体制备方法有熔融法、溶剂法和溶剂-熔融法。本研究是利用一种不同于传统制备方法的反溶剂共沉淀法制备灰黄霉素固体分散体,以期弥补灰黄霉素水溶性差、生物利用度低的缺陷,增加临床用药选择性,为提高灰黄霉素溶解性的开发研究提供新思路。

图1 Gri (a) 和HPMCAS (b) 的结构式
1 材料与方法
1.1 试验材料与动物灰黄霉素标准品98%(上海源叶生物科技有限公司);灰黄霉素原料药97%(美国葛兰素史克公司);HPMCAS(HF、MF、LF 3种规格)购于日本信越株式会社;N,N-二甲基甲酰胺 (DMF) 购于天津市科密欧化学试剂有限公司;健康成年雄性比格犬6只,体质量为12~16 kg(购于北京玛斯生物技术有限公司),试验前适应性饲养7 d,将比格犬随机分为2组,每组3只,试验前禁食12 h,可自由饮水。
1.2 灰黄霉素反溶剂的选择配制不同pH的水溶液,各取6 mL后加入50 mg灰黄霉素,100 r/min,37℃ 震荡24 h,过滤取上清,用紫外分光光度计在296 nm处测定吸光度代入标曲,选出灰黄霉素溶解度最低的水溶液。
1.3 灰黄霉素固体分散体的制备分别取1 g HPMCAS-HF、HPMCAS-MF、HPMCAS-LF加入到10 mL的N,N-二甲基甲酰胺中充分搅拌。将适量灰黄霉素按照5∶5,4∶6,3∶7,2∶8,1∶9加入到不同规格的聚合物溶液中,充分混合后匀速注入反溶剂中,抽滤得沉淀,冻干后研磨过筛备用。
1.4 灰黄霉素固体分散体的表征
1.4.1粉末X射线衍射 (PXRD) 分别将Gri、3种不同规格聚合物及共沉淀产物过筛后平铺于样品槽内进行测试。测试条件为:电压40 kV,电流15 mA,测试步长为0.01°,扫描速度为20°/min,扫描范围为3~40°,测试温度为室温。
1.4.2差示量热扫描 (DSC) 分别将3~5 mg待测样品研磨过筛干燥去水后放置于标准铝盘密封,铝盘盖扎孔,测试时空铝盘为对比参照,另一个铝盘装待测样品。测试条件为:采用N2气氛,升温速率为10℃/min,温度范围为25~250℃,N2流速为50 mL/min。
1.4.3红外光谱分析 (FT-IR) 样品制备:采用常规压片的方法,先将溴化钾研磨过筛成2 μm以下的粉末,干燥备用。按照样品和溴化钾1∶100的比例,取2 mg的待测样品和溴化钾粉末于研钵中,研磨混合均匀,压片后测定。测试条件为:波数范围4 000.0~400.0 cm-1,分辨率为0.1 cm-1。
1.4.4扫描电子显微镜 (SEM) 将待测样品粉末黏到导电胶上,控制激发电压为20 kV,在真空条件下将待测样品表面喷金后测试观察样品的外观及形态。
1.5 灰黄霉素固体分散体的体外溶解度测定
1.5.1粉末溶出 分别取Gri原料药200 mg、3种规格载体的固体分散体及对应的物理混合物(均含原料药200 mg)于烧杯中,分别加入500 mL的pH 6.8缓冲盐,温度为 (37.0±0.5)℃,转速为250 r/min。分别在0.5,1.0,2.0,5.0,10.0,15.0,20.0,30.0,45.0,60.0,90.0,120.0,150.0,180.0和240.0 min取出500 μL并补充等温等量的溶出介质,立即用0.22 μm滤膜过滤,在296 nm波长处测定吸光度,平行3次。
1.5.2溶出残渣分析 将粉末溶出试验后的混悬液过滤,沉淀物干燥后研磨过筛(80目),进行粉末X射线衍射分析,确定溶出后样品性质是否发生变化。
1.6 稳定性研究分别称取30 mg 3种不同载体的灰黄霉素固体分散体于平皿中,置于温度为40℃,湿度为75%的药品稳定性试验箱中,并分别在0,15,30,60,90,120 d取出适量样品进行粉末X射线衍射分析。
1.7 药物代谢动力学
1.7.1给药与采样 分别将灰黄霉素与其固体分散体混悬于0.5%的羧甲基纤维素钠中,以40 mg/kg的剂量灌胃给予比格犬。分别于0.083,0.250,0.330,0.500,1.000,2.000,4.000,6.000,8.000,10.000和12.000 h于比格犬前肢静脉处采血1 mL,置于含肝素钠的离心管中,12 000 r/min离心10 min,-20℃备用。
1.7.2样品处理 取100 μL血浆样品加100 μL乙腈,涡旋振荡2 min,12 000 r/min离心10 min,取上清用0.22 μm滤膜过滤后用HPLC进行Gri的含量测定。
1.7.3高效液相色谱条件 色谱柱:SunFire®C18 (4.6 mm×250 mm);流动相:乙腈∶水=0.55∶0.45;流速:1 mL/min;柱温:37℃;进样量:20 μL;检测波长:291 nm。
1.7.4统计学分析 药物代谢动力学数据采用DAS 3.5软件进行药时曲线下面积 (AUC0~∞)、最大血药浓度(Cmax)、达峰时间 (Tmax)、半衰期 (T1/2)、滞留时间 (MRT0~∞) 等药物代谢动力学参数计算分析。采用SPSS 19.0软件对数据进行显著性差异分析,以P<0.01(差异极显著)或P<0.05(差异显著)为显著性标准。
2 结果
2.1 灰黄霉素反溶剂的选择灰黄霉素在不同的pH水溶液中的溶解度如表1所示,在pH=3中溶解度最低,因此选择pH=3作为灰黄霉素固体分散体制备时的反溶剂。

表1 灰黄霉素在不同pH水溶液中的溶解度 mg/L
2.2 灰黄霉素固体分散体的PXRD结果待测样品的PXRD结果如图2所示。由Gri的X射线衍射图可知在 (10.6±0.2),(13.1±0.2),(14.5±0.2),(16.5±0.2),(23.7±0.2),(26.6±0.2),(28.4±0.2) °处均出现窄而尖的衍射峰,证明Gri是一种高结晶性的药物[7];而HPMCAS-HF、HPMCAS-MF、HPMCAS-LF均无明显的特征衍射峰;物理混合物的衍射峰与Gri的位置相同,但峰的强度比Gri弱,表明有结晶性药物的存在;在Gri-HF ASD比例为2∶8,3∶7,4∶6,5∶5时可以明显观察到与Gri有着相同位置的晶型衍射峰,证明没有完全形成无定形固体分散体;而1∶9比例的Gri-HF ASD未观察到任何晶型衍射峰,证明该比例完全形成无定形固体分散体;而Gri-MF ASD和Gri-LF ASD与Gri-HF ASD一样,表明仅1∶9比例的Gri完全形成无定形分散于载体之中,形成了固体分散体。

图2 Gri、PM、Gri-HF ASD(Gri∶HF=1∶9,2∶8,3∶7,4∶6,5∶5)、Gri-HF ASD (Gri∶HF=1∶9)、Gri-MF ASD (Gri∶MF=1∶9)、Gri-LF ASD (Gri∶LF=1∶9)、HPMCAS-HF、HPMCAS-MF和HPMCAS-LF的PXRD图
2.3 灰黄霉素固体分散体的DSC结果各样品的DSC如图3所示。Gri在220℃有1个明显的吸热峰为Gri的熔点[8];HPMCAS-HF、HPMCAS-MF和HPMCAS-LF在温度升高过程中均未出现任何吸热峰;物理混合物在220℃处可见Gri的熔点峰,证明载体与原料药为物理混合物;Gri-HF ASD、Gri-MF ASD和Gri-LF ASD在220℃附近均未出现吸热峰,表明Gri以无定形的状态分散于载体之中,与上述PXRD结果吻合。
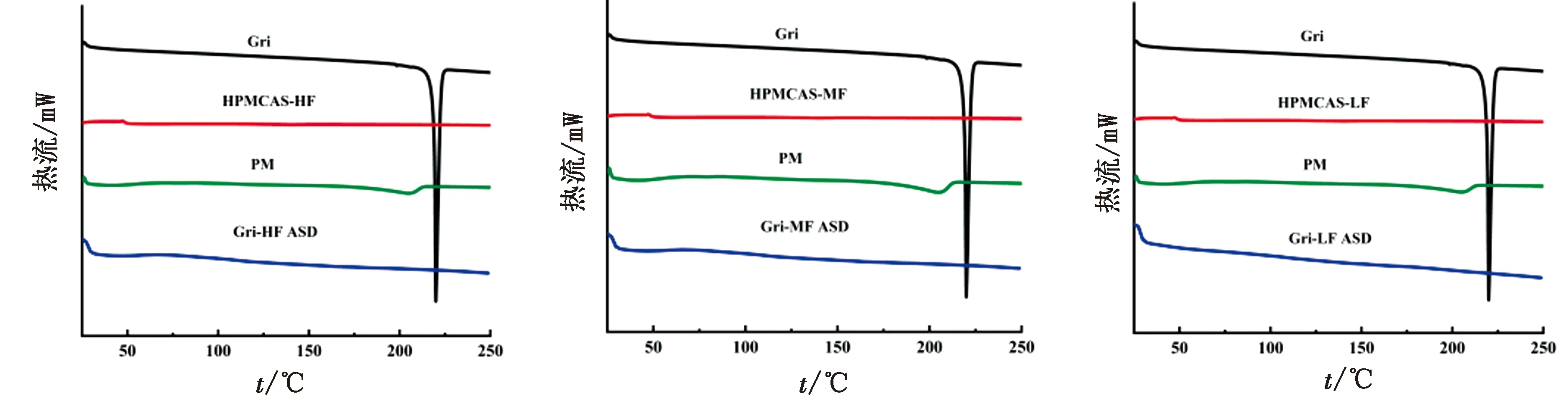
图3 Gri、PM、Gri-HF ASD (Gri∶HF=1∶9)、Gri-MF ASD (Gri∶MF=1∶9)、Gri-LF ASD (Gri∶LF=1∶9)、HPMCAS-HF、HPMCAS-MF和HPMCAS-LF的DSC图
2.4 灰黄霉素固体分散体的FT-IR结果各样品的FT-IR结果如图4所示。Gri在2 944 cm-1处的谱带为-OCH3官能团的伸缩振动峰;HPMCAS-HF、HPMCAS-MF和HPMCAS-LF在2 937和1 745 cm-1分别为-OCH3和-C=O官能团的伸缩振动峰[9];在物理混合物中既出现了载体中的2 937 cm-1(-OCH3)官能团谱带,也有Gri中的1 745 cm-1(-C=O)官能团谱带,表明Gri与载体没有发生结构变化,只是简单的物理混合;在Gri-HF ASD (Gri∶HF=1∶9)、Gri-MF ASD (Gri∶MF=1∶9)、Gri-LF ASD (Gri∶LF=1∶9)的图谱中,Gri的甲氧基峰由原来的2 944 cm-1蓝移到3 132 cm-1。推测Gri在形成固体分散体的过程中,改变了分子中的键的伸缩振动情况,表明药物与载体之间发生了氢键的结合。

图4 Gri、PM、Gri-HF ASD (Gri∶HF=1∶9)、Gri-MF ASD (Gri∶MF=1∶9)、Gri-LF ASD (Gri∶LF=1∶9)、HPMCAS-HF、HPMCAS-MF和HPMCAS-LF的FT-IR图
2.5 灰黄霉素固体分散体的SEM结果各样品的SEM结果如图5所示。Gri是具有短而钝且不规则的棒状晶体结构;HPMCAS-HF、HPMCAS-MF和HPMCAS-LF的形状具有相似性,都是不规则的块状结构;Gri-HF ASD (Gri∶HF=1∶9)、Gri-MF ASD (Gri∶MF=1∶9)、Gri-LF ASD (Gri∶LF=1∶9) 中没有明显的棒状晶体结构,出现了少许球状颗粒及微小聚集体分布在蜂窝状的空腔内,且呈疏松多孔状的结构,表明Gri以无定形的状态分散于载体的孔穴中。

图5 Gri、Gri-HF ASD (Gri∶HF=1∶9)、Gri-MF ASD (Gri∶MF=1∶9)、Gri-LF ASD (Gri∶LF=1∶9)、HPMCAS-HF、HPMCAS-MF和HPMCAS-LF的SEM图
2.6 灰黄霉素固体分散体最佳比例的体外溶出结果
2.6.1粉末溶出结果 Gri、PM、Gri-HF ASD (Gri∶HF=1∶9)、Gri-MF ASD (Gri∶MF=1∶9)、Gri-LF ASD (Gri∶LF=1∶9) 在37℃、pH 6.8磷酸盐缓冲液条件下的溶出结果如图6所示。从图中可以看出Gri达到平衡时的溶解度是6 mg/L[10]。3种PM均在30 min达到平衡状态,平衡时的溶解度分别为12 mg/L (Gri-HF)、7 mg/L (Gri-MF和Gri-LF),分别是Gri原料药的2.0和1.2倍。Gri-HF ASD的溶出曲线呈先升后降的趋势,在30 min前呈上升趋势,30 min后开始下降,最高点时的溶解度为113 mg/L,是Gri原料药的18倍,最终下降到49 mg/L,是Gri原料药的8倍。Gri-MF ASD与Gri-LF ASD的溶出曲线类似,均在2 min后开始下降,但Gri-LF ASD下降的趋势较Gri-MF ASD迅速,最高点时的溶解度分别为205 和132 mg/L,分别是Gri的34和22倍,下降到最低点时的溶解度分别为23 和28 mg/L,是Gri的4倍左右。从溶出曲线可以看出该方法制备出的灰黄霉素无定型固体分散体明显改善了灰黄霉素的溶解度。
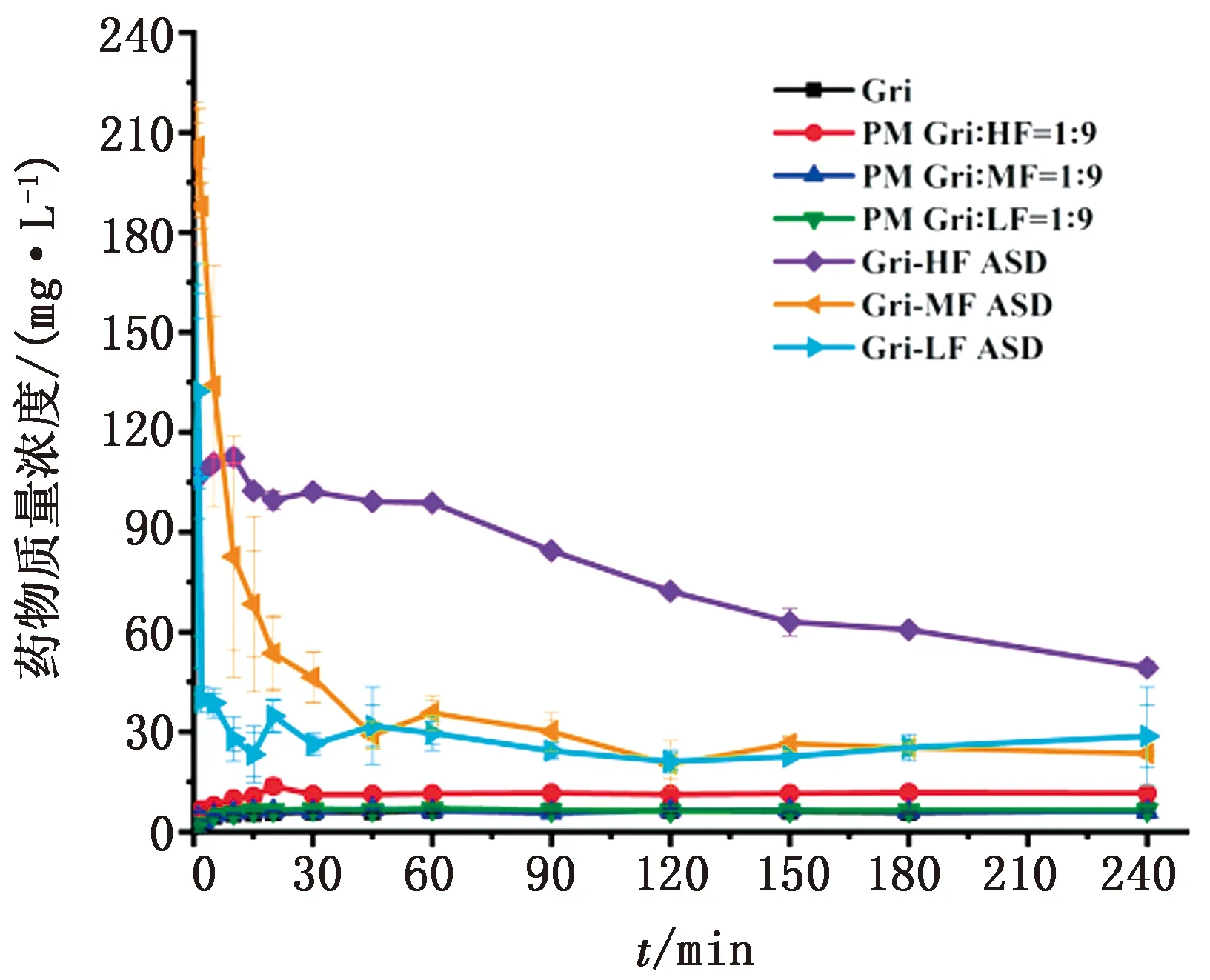
图6 Gri、PM、Gri-HF ASD、Gri-MF ASD、Gri-LF ASD的体外溶出曲线图
2.6.2溶出残渣X射线衍射结果 Gri、Gri-HF ASD (Gri∶HF=1∶9)、Gri-MF ASD (Gri∶MF=1∶9)、Gri-LF ASD (Gri∶LF=1∶9) 在37℃、pH 6.8缓冲盐条件下溶出后残渣的PXRD结果如图7所示。Gri-HF(1∶9)的溶出残渣在(28.4±0.2)°出现了灰黄霉素的晶体衍射峰,Gri-MF(1∶9)和Gri-LF(1∶9)的溶出残渣在的溶出残渣在(23.7±0.2)°出现了灰黄霉素的晶体衍射峰,表明灰黄霉素固体分散体在水溶性介质中有一部分从无定形状态转为了晶体状态。

图7 Gri、Gri-HF ASD (Gri∶HF=1∶9)、Gri-MF ASD (Gri∶MF=1∶9)、Gri-LF ASD (Gri∶LF=1∶9) 溶出残渣的PXRD图
2.7 稳定性试验结果在加速稳定性的试验条件下(温度为40℃,湿度为75%),灰黄霉素固体分散体在不同时间点的X射线衍射图和差示量热扫描结果如图8所示。在不同时间点的X射线衍射图中均无任何晶型的衍射峰,证明用反溶剂共沉淀法制备出的固体分散体在高温高湿的环境下稳定性良好。

图8 Gri-HF ASD (Gri∶HF=1∶9) (a)、Gri-MF ASD (Gri∶MF=1∶9) (b)和Gri-LF ASD (Gri∶LF=1∶9) (c)稳定性的PXRD图
2.8 药物代谢动力学结果根据体外溶出试验结果,选择Gri、Gri-HF ASD (Gri∶HF=1∶9) 在比格犬进行口服药物代谢动力学研究。药物代谢动力学结果如图9和表2所示。Gri-HF ASD的达峰时间Tmax从0.67 h缩短到到0.39 h,表明Gri-HF ASD在比格犬体内吸收迅速;2组的最大血药质量浓度Cmax分别为0.23 和1.43 mg/L,固体分散体组比原料药组提高了6.2倍;2组的AUC分别为0.84 和1.90 mg/(L·h-1),固体分散体组比原料药组提高了2.3倍。
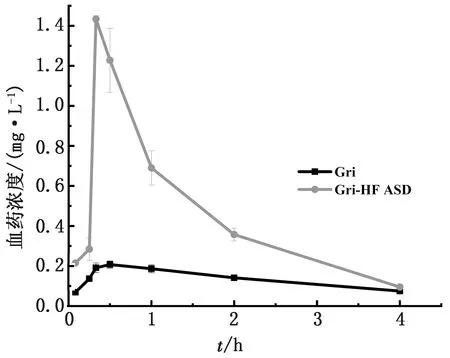
图9 口服Gri和Gri-HF ASD在比格犬体内的血药浓度-时间曲线

表2 比格犬口服Gri和Gri-HF ASD主要药 动学参数对比 (n=3)
3 讨论
固体分散体 (SDs) 在改善药物溶解度相比其它策略受到了更多的关注,与传统策略相比,因其颗粒尺寸的减小、团聚的减少和润湿性的改善提高了药物的生物利用度[11]。而因为聚合物能抑制药物的结晶,一方面有助于药物的长期储存稳定性;另一方面,有助于维持一个理想的过饱和水平,因为它阻止了溶剂介导的药物重结晶[12]。常用的固体分散体制备方法包括熔融法、溶剂法、熔融-溶剂法等。熔融法具有时间短、无溶剂、成本低及抗毒性等优点,但制备过程中高温的使用,药物的分子迁移率加快,有些药物会在熔融过程发生化学降解或者升华。溶剂法的优点为避免高热,适用于对热不稳定或挥发性药物。但使用有机溶剂的用量较大,成本高,且有时有机溶剂难以完全除尽[13]。溶剂-熔融法优点是药物受热时间短、稳定,产品质量好,缺点是仅限于小剂量药物[14]。
新型共沉淀技术运用溶剂-反溶剂原理和经典成核理论,该方法是基于含溶质的有机溶液与可混溶的反溶剂的混合,在快速混合的条件下产生高的过饱和状态促使快速成核从而瞬时产生沉淀[15],使药物以无定形状态均匀分布于载体之中,该方法操作简单成本低对环境污染小。CHOW等[16]采用共沉淀法制备了姜黄素无定形固体分散体,并比较了混合速度以及溶剂扩散系数对其粒径的影响,证明了该方法制备出的固体分散体粒径小且稳定性良好。本研究是采用新型共沉淀的方法制备了灰黄霉素固体分散体,其饱和溶解度提高了8倍,Cmax提高了6.2倍。
HPMCAS为肠溶性载体,在pH 6.0条件下溶解,具有两亲性结构,既有亲水基团又有疏水基团[17-18]。HPMCAS因官能团的含量不同分为3种不同型号。HPMCAS-HF含有较多的疏水性取代基乙酰基,当作为聚合物时与药物的相互作用为较强的疏水作用,从而产生较低的药物过饱和度,因此药物溶出度较低,但能够较好的维持过饱和度。而HPMCAS-MF和HPMCAS-LF含有较多的亲水取代基琥珀酰基,因此与水有较强的相互作用,药物溶出度较高,但维持过饱和的能力较弱因为与水的作用力较弱从而减缓药物结晶[19]。因此LF和MF的重结晶速度快,溶解度下降迅速,本研究中利用LF和MF为载体的固体分散体均在2 min后溶解度急剧下降,分别从205 和132 mg/L下降至23和28 mg/L;而HF的重结晶速度缓慢,溶解度下降缓慢,本研究中HF在30 min后开始缓慢下降,最终从113 降至49 mg/L。由于HF在胃肠道能够保持更长时间过饱和从而减缓灰黄霉素在胃肠道吸收过程中的重结晶速度[20-22],因此以HF在聚合物的固体分散体的生物利用度比灰黄霉素提高了2.3倍。
灰黄霉素是一种高结晶倾向的药物[23],当载体材料HPMCAS在pH6.8的缓冲盐介质中部分溶解后,无定形灰黄霉素从载体中释放暴露于水溶性介质中后发生快速和广泛的结晶[24-25],这是由于药物与聚合物的相容性和相互作用[26-27],ASD中的无定形药物在溶解介质中与水接触时可能发生相分离和重结晶[28-30]。因此在溶出后的残渣PXRD图中可以看出晶型物质的出现。而由于LF和MF含有较多的亲水取代基琥珀酰基,具有较高的聚合物润湿性,导致非晶态灰黄霉素的初始释放速度比HF快,因此在溶出开始时LF和MF的最高浓度比HF高,同时由于溶解速度快与水溶性介质接触时间早导致快速结晶[31-32]。因此可以从溶出残渣PXRD图中观察到LF和MF较HF的重结晶性高。
猜你喜欢
昆钢科技(2021年3期)2021-08-23
煤质技术(2021年3期)2021-07-07
小读者·阅世界(2020年5期)2020-06-01
中国材料进展(2020年4期)2020-05-23
火工品(2019年3期)2019-09-02
数学大王·趣味逻辑(2019年8期)2019-08-27
故事会(2009年8期)2018-09-03
浙江农业科学(2017年4期)2017-05-11
湖北农业科学(2016年5期)2016-10-19
餐饮世界(2015年8期)2015-06-27
