
中国福建地区H5Nx水禽流感病毒Clade 2.3.4.4分支分子特性分析
2023-01-31 07:30朱春华陈翠腾施少华刘斌琼傅光华
中国兽医学报 2022年11期
朱春华,陈翠腾,陈 珍,施少华,刘斌琼,傅光华,黄 瑜
(福建省农业科学院 畜牧兽医研究所,福建 福州 350013)
高致病性禽流感(highly pathogenic avian influenza,HPAI)是由H5亚型或其重组毒株(包括H5N2、H5N5、H5N6和H5N8)流感病毒引起的一种禽类的烈性传染病,引起家禽和野生鸟类数千次禽流感暴发并造成大量死亡,对世界养禽业造成了巨大经济损失。目前H5N1 Gs/GD谱系HPAI病毒已进化为10个基因不同的病毒进化支(Clade 0~Clade 9)和多个亚进化支[1],其中Clade 2谱系为主要分支,自2008年以来,在中国的家禽中,尤其是家鸭和活禽市场的家禽中发现了具有Gs/GD谱系H5进化分支Clade 2.3.4遗传骨架的HPAI亚型H5N2、H5N5和 H5N8,这些亚型随后进化成不同的子分支,其中最为醒目的是Clade 2.3.4.4亚分支[2-3]。HPAIV Clade 2.3.4.4与 H5N1亚型病毒的其他进化支及本地LPAIV亚型进行了基因重组[4],不同Clade 2.3.4.4(a、b、c和e)的病毒在抗原性上往往不同。多种禽类,包括野生和家养水禽、甚至动物园的鸟类,似乎都可以感染和/或传播HPAIV进化支Clade 2.3.4.4。与以前的H5N1 HPAIV相比,这些重组病毒在鸟类中表现出致病性的改变。Clade 2.3.4.4进化分支病毒的致病性不仅在宿主之间不同,而且在同一种宿主内也不同[5]。
不同流感病毒之间的重组被认为是新病毒出现的主要机制,野生鸟类、家鸭体内几种H5Nx流感病毒的共同传播促成了中国南方禽流感病毒的抗原多样性和遗传多样性[6]。中国南方是家养水禽的主要养殖区之一,家禽常与野生水鸟共享水域。2010年,中国东部种鸭中发现了H5N8和H5N5的重组病毒[7-8],警示病毒的基因重组可能导致新的流感病毒亚型的产生。2014年浙江、湖北和广东养殖场出现了H5N6禽流感,2015年湖南和江苏也出现同一亚型的病例[9]。近3年来H5N8流感和相关病毒在野鸟感染和家禽中频繁暴发,HPAIV常见的H5基因片段易与其他流感病毒重组[10-11]。因此,H5Nx是新的致病变异毒株的持续来源。禽流感Clade 2.3.4.4分支HA基因在家禽体内循环数周或数年后,基因发生特定突变,具有明显的杂交性,从而产生了具有多种神经氨酸酶亚型的重组体。
该研究旨在对福建省病死家禽及养禽场采集的组织样品和/或棉拭子样品、活禽市场及其排污沟采集的棉拭子样品分离的H5Nx流感毒株、以及东南地区主要流行毒株、迁徙鸟类、国内代表性毒株等的血凝素基因序列进行遗传进化分析,探讨Clade 2.3.4.4分支病毒在遗传演化过程中亚型的转变,进一步丰富我国福建地区禽流感病毒的分子流行病学,为福建省禽流感的预警预报及其防控决策提供科学依据,为保障福建省乃至我国养禽业持续健康发展、禽产品有效供给及公共安全、社会稳定发挥重要的作用。
1 材料与方法
1.1 毒株和来源试验所涉及的福建及周边省份禽流感H5Nx毒株或样品如下: A/chicken/China/FJ-XZ-18-87/2015(H5N8)(简称FJ1519)、A/chicken/Chian/FJ-XZ-18-27/2016(H5N6)(简称FJ1682)、A/Pigeon/Fujian/1.17_FZHX0104-O/2017(H5N6)、A/Goose/Fujian/3.15_FZHX0008-C/2018(H5N6)、A/Muscovy duck/China/H5N6/2020(H5N6)、A/goose/Fujian/11.3_FZHX1102-C/2016(H5N6)、A/goose/Fujian/10.26_FZHX0014-O/2017(H5N6)、A/goose/Fujian/3.15_FZHX00-07-C/2018(H5N6)、A/duck/Jiangxi/2.28NCNP-23K3-OC/2018(H5N6),其中FJ1519、FJ1682来源于福建活禽市场棉拭子样品。GenBank下载国内不同时期的代表株,以及2011—2021年中国东南地区以及周边日本、韩国等H5Nx毒株(H5N1、H5N2、H5N5、H5N6和H5N8亚型),对位于同一进化分支的相同年份的毒株进行删除。
1.2 HA基因RT-PCR扩增将病料匀浆液8 000 r/min离心5 min,取250 μL上清与750 μL TRIzol混合,再加入200 μL预冷的氯仿,充分混匀后冰浴5 min,随后12 000 r/min离心15 min,取上清加入等体积的异丙醇,混匀,-20℃沉淀1 h。12 000 r/min离心15 min,弃上清,用DEPC处理过的水重悬,-20℃ 保存。用流感的反转录引物Uni-12,按照AMV反转酶说明书进行反转录合成cDNA。PCR扩增以cDNA为模板,反应体系为50 μL,反应体系为:10×buffer 5 μL,dNTP mixture (2.5 nmol/L)4.0 μL,Ex-Taq DNA polymerase 0.5 μL,引物(20 μmol/L)各1.0 μL,模板cDNA 1 μL,ddH2O 37.5 μL。反应条件如下:94℃预变性5 min;94℃ 30 s,55℃ 35 s,72℃ 2 min,进行35个循环;72℃延伸10 min。取5 μL 产物经1%琼脂糖凝胶电泳检测扩增结果。
1.3 HA基因遗传进化分析PCR扩增得到的HA基因PCR产物经切胶回收试剂盒纯化后与pMD-18T载体连接,连接反应体系如下:PCR纯化产物 50 ng,pMD-18T载体 1 μL,ddH2O补充到5 μL转化DH5α感受态细胞,PCR法鉴定重组质粒,阳性质粒送大连宝生物公司测序。应用Lasergene软件包Seqman软件进行序列拼接。NCBI网站上批量下载带FASTA格式的HA基因序列及其对应的氨基酸序列,应用MEGA-X软件中ClustalW对48株病毒HA序列进行同源序列比对,邻近归并法构建遗传进化树(Bootstrap = 1 000)[12]。Lasergene软件包中 MegAlign进行氨基酸同源性比较。
1.4 同源建模和进化保守性分析基于同源序列比对结果,选取其中1个毒株FJ1519进行同源建模。将FASTA格式的FJ1519 HA序列上传到SWISS-MODEL在线软件进行同源建模[13],选择鸭源毒株A/mallard/Vietnam/3/2003(H5N1)(PDB ID:6PCX)为模型(FJ1519与其同源性为92.66%,符合同源建模的条件),建模得到的FJ1519毒株HA蛋白结构通过PyMOL和Photoshop软件作图。通过Consurf在线软件进行同源进化分析,选择国际通用的1968年大流感的代表毒株A/Aichi/2/1968(H3N2)对HA序列排序[14],选择比对的序列为500条,选择的蛋白数据库为UNIREF-90,计算方法选择贝叶斯(Bayesian)算法,同源多序列比对选择的方法为ClustalW。
2 结果
2.1 HA基因同源进化分析通过BLAST在线分析,FJ1519毒株HA基因序列与A/goose/Eastern China/CZ/2013(H5N8)HA基因序列同源性最高,为99.00%;FJ1682与A/duck/China/FJ1602/2016(H5N6)同源性最高,为99.65%。通过HA核苷酸序列遗传进化分析发现,FJ1519和FJ1682分别属于2个不同的大分支,其中FJ1519与A/mallard/Shanghai/SH-9/2013、A/Von Schrenck's bittern/Jiangxi/Y9/2014处于同一进化分支,FJ1682与A/whooper swan/Hokkaido/X12/2017处于同一进化分支。FJ1519、FJ1682与福建省近5年分离得到的H5Nx毒株亲缘关系比较近,同源性分别为93.10%~96.10%,94.50%~99.30%;与福建省2011年从野鸭上分离的毒株A/wild duck/Fujian/1/2011(H5N1)亲缘关系比较远,同源性分别为91.90%,91.30%;与国内的H5亚型代表株A/Goose/Guangdong/1/96(Clade 0)亲源关系比较远,同源性分别为92.40%,91.30%。A/Muscovy duck/China/H5N6/2020(H5N6)毒株与FJ1519株、FJ1682株同源性分别为93.10%,94.50%;与A/Goose/Fujian/3.15_FZHX0008-C/2018(H5N6)、A/goose/Fujian/3.15_FZHX0007-C/2018(H5N6)毒株同源性最高,均为97.90%;与天鹅源的毒株A/Whooper swan/Mongolia/25/2020(H5N6)、A/Whooper Swan/Khuvsgul/#1/2020(H5N6)同源性均为97.00%,并处于同一小进化分支上,与天鹅源的日本毒株A/whooper swan/Miyagi/0402B001/2021(H5N8)同源性最低,为91.20%。从图1可知,近10年来在我国东南地区流行的H5Nx毒株主要有H5N6和H5N8亚型,且这2种亚型位于不同的大聚类分支上(图1)。本研究发现福建省分离的FJ1519毒株HA蛋白与从野鸭上分离得到的A/mallard/Shanghai/SH-9/2013(H5N8)同源性最高,为99.50%,说明这2个毒株来源于共同的祖先;江西省麻鸭上分离的A/Von Schrenck's bittern/Jiangxi/Y9/2014(H5N8)与A/mallard/Shanghai/SH-9/2013(H5N8)同源性高达99.60%,这3株病毒处于同一小进化亚分支上。A/mallard/Shanghai/SH-9/2013(H5N8)分离地上海位于跨越中国、朝鲜、日本和韩国的东亚候鸟迁徙路线上,是监测中国流感病毒的理想地点,从野鸭身上分离的新型重组H5N8 禽流感病毒通过候鸟在东亚飞行路线中传播,韩国暴发的H5N8疫情支持了这种候鸟迁徙观点的可能性[15]。此外,通过遗传进化分析,本研究也发现2020年广东省鹅源A/goose/China/21FU-008/2020(H5N8)以及2021年山东省疣鼻天鹅分离得到的A/Mute Swan/China/Shandong1/2021(H5N8)与来自日本的A/whooper swan/Fukushima/0701B002/2021(H5N8)、A/whooper swan/Miyagi/0402B001/2021(H5N8)这2个毒株处于同一进化亚分支上,说明这几株病毒HA基因来源于共同的祖先,表明东南地区鸟类的迁徙可能引起不同流感病毒之间的重组,并在病毒传播过程中发挥重要的作用[10,16]。

·.本研究所涉及的福建分离株
2.2 HA氨基酸序列分析本研究所涉及的中国东南地区Clade 2.3.3.4分离株HA序列氨基酸裂解位点主要有RERRRKR↓G、REKRRKR↓G 2种形式,较为特别的是A/Goose/Guangdong/1/1996(H5N1)裂解位点为RERRRKKR↓G、A/Goose/Eastern China/1112/2011(H5N2)裂解位点为RGKRRKR↓G,所有Clade 2.3.3.4分支HA蛋白裂解位点均为连续的碱性氨基酸。A/wild duck/Fujian/1/2011裂解位点为IEKRRKR↓G,FJ1519裂解位点为REKRRKR↓G,其他裂解位点为RERRRKR↓G,上述这些毒株HA蛋白裂解位点均符合HPA1特征(表1)。

表1 H5Nx流感毒株HA蛋白序列关键氨基酸位点
以FJ1519毒株为例,HA核苷酸序列长度为1 704 bp,编码568个氨基酸,通过在线软件SignalP和TMHMM预测发现,HA序列全长N端含有信号肽(16个氨基酸),C端含有1个跨膜结构域(23个氨基酸),其中17~531位氨基酸为胞外可溶性区段。进一步通过氨基酸进化保守性分析,结论发现多数氨基酸位点在遗传进化上是保守的(绛紫色),但有一些位点较为灵活(海蓝色),表明不同的H5Nx毒株之间存在抗原差异(图2)。通过Bioedit同源序列比对,发现研究中所涉及中国福建地区分离株HA氨基酸序列均含有6个糖基化位点,氨基酸基序分别为NSTE(或NSTK)、NVTV、NNTN、NPTT(或NPDT)、NSSM、NGTY和NGSL,其中第209位氨基酸由于其后1位氨基酸为Pro,在天冬酰胺之后的脯氨酸在多数情况下会使天冬酰胺无法接近来阻止N-连接的糖基化。因此,NPTT(或NPDT)这个位点需要进一步证实。
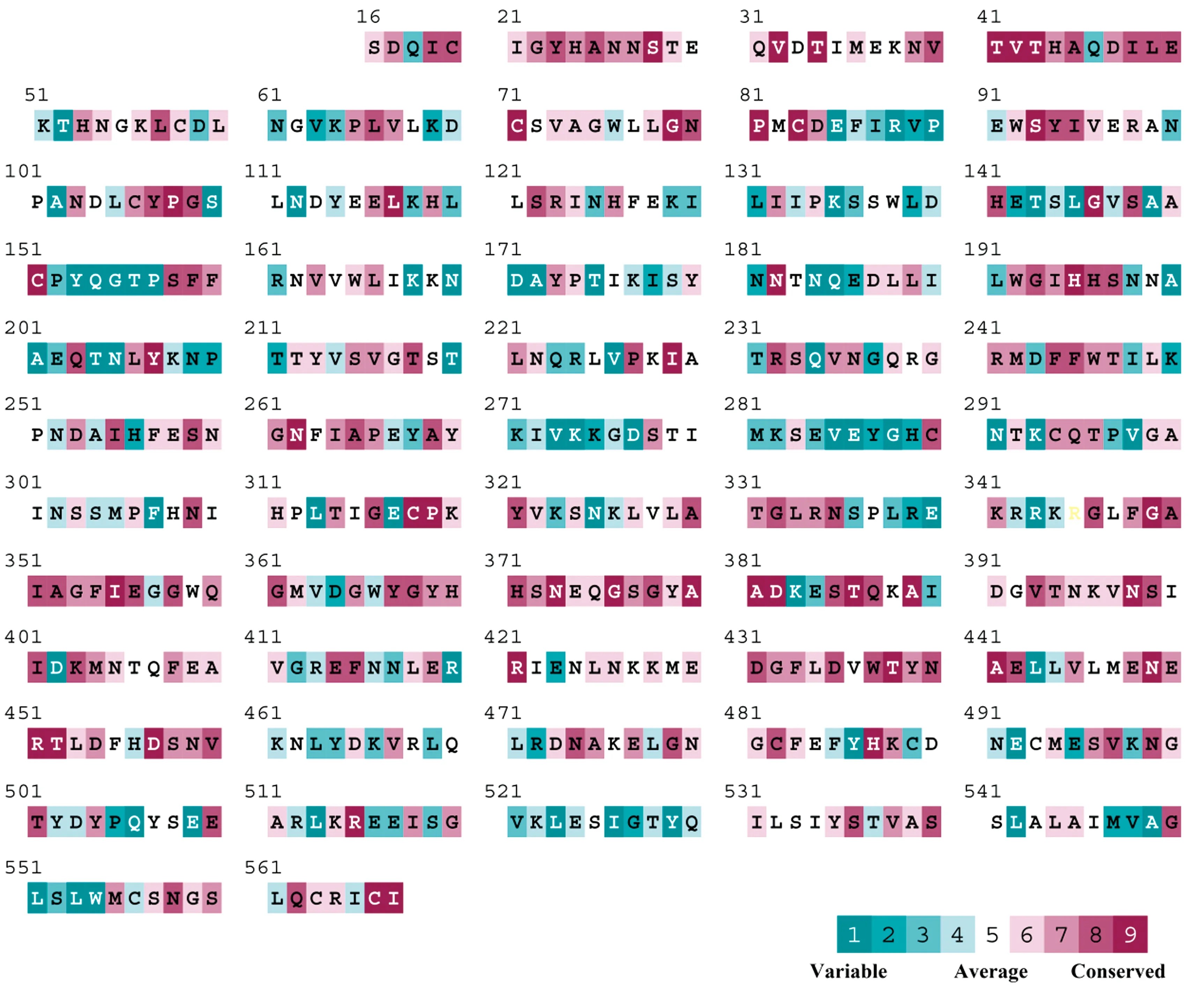
图2 HA蛋白氨基酸进化保守性分析
2.3 病毒分子特征流感病毒受体结合特点与宿主嗜好密切相关,对唾液酸受体的结合偏好性研究是了解受体结合位点突变的重要领域,人流感病毒HA偏好识别α-2,6-唾液酸(人源受体),而禽流感病毒HA优先识别α-2,3-唾液酸(禽源受体)[17-18]。通过HA同源序列和二级结构比对,研究发现H5Nx亚型Clade 2.3.4.4受体主要结合位点由Y98、W153和H183这3个氨基酸残基组成(以H3排序),侧壁由130-loop(A138)、190-螺旋(E190、L194、Y195)和220-loop(G225和G228)这3个二级结构组成[17,19],结合以往研究报道与受体结合相关的关键氨基酸位点Y98、W153、H183、A138、E190、L194、Y195、G225、Q226和G228分别对应于FJ1519毒株上Y106、A150、W165、H195、E202、L206、Y207、G237、G240,这几个氨基酸位点在遗传进化上高度保守(图2,3)。从表1可知,第193和227位氨基酸在遗传进化过程中易发生变异,从福建省分离得到的毒株上可知第193位氨基酸为R、D、N、K,227位点主要为S、Q、R,福建省分离得到的H5Nx禽流感病毒Clade 2.3.3.4 HA序列具有典型的禽类受体结合特征,尤其是,第226氨基酸均为谷氨酰胺(Q),可赋予其结合到禽类受体的特性,优先识别禽源α-2,3唾液酸[15,17]。禽源H5Nx Clade 2.3.3.4 HA序列第226位均为Q,人源代表株 A/Aichi/2/1968(H3N2)该位点为L,当禽流感毒株HA序列中该位点从Q突变成L时,即可发生受体结合特性的转变,从偏好结合禽源受体变成人源受体[17]。此外,HA上L226V和V226I突变,也会影响到病毒受体的结合能力发生变化。

虚线框代表的是受体结合区域;棍状形式表示受体结合区氨基酸位点
3 讨论
近年来,来自Gs/GD谱系重组的H5Nx HPA-IV相继在许多国家暴发,通过迁徙水鸟的传播在家养和野生鸟类中引起了多次洲际疫情,并对野生鸟类和家禽造成了重大影响,对养禽业和人类健康构成巨大的威胁[20-21]。自2014年以来,HPAI进化支Clade 2.3.4.4病毒通过野生鸟类在全球范围内迅速传播,并通过与当地流行的LPAIV重新组合而进化。多种水禽,包括野生和家养水禽,甚至动物园鸟类,似乎都可以感染和/或传播Clade 2.3.4.4进化支的病毒。尽管一些研究指出,与亲本Gs/GD(H5N1)相比,这些H5Nx重组株的毒力降低,但一般来说Clade 2.3.4.4感染的水禽仍表现出典型的HPAI病毒感染的临床疾病、死亡率和病理特征[22-23]。研究前期分子流行病学研究表明H5N6、H5N8亚型是目前主要流行的H5Nx病毒亚型[2],遗传进化分析发现这2种亚型在中国东南地区、中国台湾地区、日本、朝鲜之间流行。不同流感病毒之间的重组被认为是新病毒出现的主要机制,H5Nx型Clade 2.3.4.4亚分支流感病毒在野鸟、家禽的共循环导致了我国南方地区流感病毒的抗原性和基因多样性。
这项研究中H5Nx毒株具有典型的禽类受体结合特征,而前期流行病学调查中,发现福建省禽类群体中不断出现新的重组和变异流感毒株,多数 H9亚型(H9N2、H9N6)禽流感病毒以及H7N9和H6N2毒株倾向于结合人源受体,这增加了人感染禽流感的风险[2]。本研究中所涉及的毒株受体结合特征为禽源受体,第160位氨基酸均为丙氨酸(A),以往研究发现H5Nx亚型病毒Clade 2.3.4.4分支HA蛋白上该位点具有双受体结合特征,GAO等[24]发现H5N6亚型毒株中T160A突变体导致该毒株158位糖基化位点缺失,并影响到毒株致病性,从而引起宿主免疫应答的改变。H5Nx毒株HA蛋白T160A取代或158位点去糖基化使得毒株获得双重受体结合特性,这可能是Clade 2.3.4 H5N1进化到Clade 2.3.4.4 H5Nx 的重要分子标记,可作为评价分离株大流行的关键指标。与野生型毒株相比,S221P突变的H5N1毒株显示出有限的双重受体特异性;S221P与结合位点的K216E突变协同作用导致毒株与人源受体α-2,6-唾液酸的结合亲和力增加[25]。总之,关键氨基酸受体结合位点突变导致受体亲和力改变,并对病毒生物学功能比如感染和传播效率产生影响,因此必须加强流感病毒HA蛋白上这些关键氨基酸位点的监测。
HA蛋白是A型流感病毒主要表面抗原和保护性抗原,是流感病毒重要的毒力因子,近年来研究专注于病毒上关键氨基酸位点改变对病毒毒力和致病性的影响。WU等[26]将A/duck/Zhejiang/W24/2013(H5N8)毒株连续在小鼠的肺-肺传代,发现HA (A149V) 和 PB2 (E627K) 蛋白的突变导致感染小鼠后毒力增强,且病毒表现出组织嗜性范围变广,提示这2个突变体是H5N6在哺乳动物宿主体内适应和增强致病性的关键因素。YU等[27]发现水禽源H5N5亚型Clade 2.3.4.4进化支病毒在小鼠体内连续传代后可改变小鼠毒力,小鼠适应变异毒株HA分子茎区F430L(H3排序)替代的病毒的MLD50(mouse lethal dose)降低了3.16倍,表明HA-F430L突变体对于H5N5水禽流感病毒对哺乳动物的适应性很重要。这项研究涉及的H5Nx亚型毒株Clade 2.3.4.4第430位氨基酸均为L,这些发现强调了持续监测家禽是否含有这些氨基酸取代位点的H5Nx亚型AIV显得尤为重要,表明传代获得的氨基酸变异的信息可以为宿主提供特异性变化的候选位点,为评估新出现毒株的风险提供参考。
通过氨基酸同源序列比对分析发现不同毒株之间存在抗原差异,但前期实验室分离毒株免疫血清与Re-8标准抗原有较高血凝抑制价,Re-8标准血清与分离毒株有较高血凝抑制价,表明分离毒株与Re-8疫苗抗原性差异不明显(另文发表),商品化的流感疫苗目前仍适用,但实时监测商用流感疫苗和目前流行的病毒之间存在氨基酸、抗原差异仍然是流感监测环节的关键[28]。疫苗接种与未接种的国家之间,H5Nx病毒的进化动态存在差异,与从未使用过疫苗接种的国家中传播的病毒相比,H5N1禽流感疫苗接种的国家中传播的病毒种群的进化率和阳性选择位点的数量都更高[29],表明疫苗接种不足对病毒进化具有潜在影响。此外, H5Nx亚型病毒Clade 2.3.4.4进化分支的广泛宿主范围(鸡、鸭、鹅、鸽、鹌鹑、天鹅、秋鳽等)可能导致无法识别的病毒传播,这也很好解释了该谱系最近成功的全球化。最近在欧洲和北美家禽养殖场引起严重疫情的 H5N8 病毒已经证实来自于在北极繁殖地相遇的迁徙鸭、天鹅和鹅[11]。本研究也发现病毒重组与传播与洲际野生鸟类迁徙密切相关,新型重组病毒的出现及其在鸟类种群中的传播备受关注。近年来中国水鸟数量显著增加,中国被认为是全球水鸟携带流感病毒的最大宿主栖息地[6]。这些新型H5N6、H5N8病毒的共同传播对家禽业、对人类健康均造成了巨大的威胁[22,30],因此,将来应加强对福建省湿地迁徙鸟类的流感监测。
禽流感是由禽类宿主生态、病毒特征和环境变量之间错综复杂的相互作用形成[31],它在人类宿主中有效感染和传播的潜能是一个重大的全球公共健康问题[15]。野生鸟类的迁徙在病毒传播过程中发挥重要的作用,因此,对中国东南重要的野生鸟类聚集区和家禽进行长期和系统的流感监测,并提供有价值的流行病学信息,有助于对家禽乃至人类健康的威胁提供早期预警,能够对新出现的H5Nx病毒的威胁起到良好的警示作用。此外,应根据抗原差异优化疫苗配方和疫苗运载系统以提高疫苗效力,这将为预防新型禽流感的暴发和评估人畜共患病的可能性提供参考。
猜你喜欢
广东农业科学(2022年11期)2023-01-13
黑龙江大学自然科学学报(2022年1期)2022-03-29
科学大观园(2022年2期)2022-01-23
文萃报·周二版(2021年47期)2021-12-14
数学物理学报(2021年4期)2021-08-30
世界科学技术-中医药现代化(2021年12期)2021-04-19
学生天地(2019年28期)2019-08-25
猪业科学(2018年6期)2018-01-23
中国猪业(2017年11期)2017-12-11
中国医药科学(2015年15期)2015-02-27
