
甲泼尼龙关键中间体的合成工艺研究
2023-01-22 02:19:56林建东叶有志蒋建武
浙江化工 2022年12期
林建东,叶有志,蒋建武
(台州仙琚药业有限公司,浙江 台州 317016)
甲泼尼龙(methylprednisolone)是第2 代甾体类糖皮质激素药物,其结构式见图1。临床上主要用于炎症(如风湿性疾病、原疾病、皮肤疾病等)、免疫抑制(器官移植)、获得性溶血性贫血、成人继发型血小板减少、肿瘤(成人白血病和淋巴瘤、儿童急性白血病)、休克、内分泌失调、原发及继发性肾上皮质功能减退等多种疾病的治疗[1-2],市场前景广阔。例如,法玛西亚普强公司(Pharmacia and upjohn)研发的注射用甲泼尼龙琥珀酸钠(商品名称为甲强龙)具有显著的抗炎、免疫抑制及抗过敏活性。大剂量注射用甲强龙也可用于短期内控制某些急性重症疾病[3]。与泼尼松龙相比,甲泼尼龙除了具有糖皮质激素的药理活性外,其还有更强的抗炎作用和较弱的水、钠潴留副作用。
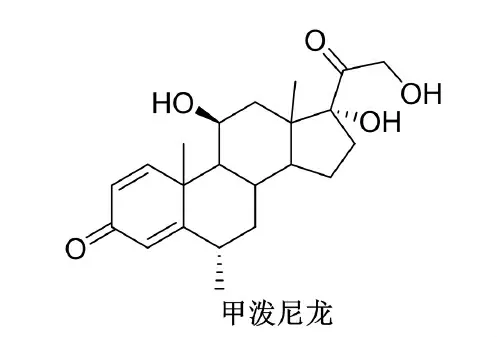
图1 甲泼尼龙的结构式
发展高效、对环境友好且易产业化的甲泼尼龙合成工艺一直是学者们的研究重点。陈伟等[4]以氢化可的松为反应起始原料,经亚甲基反应、还原反应和脱氢反应制备得到甲泼尼龙(Scheme 1)。
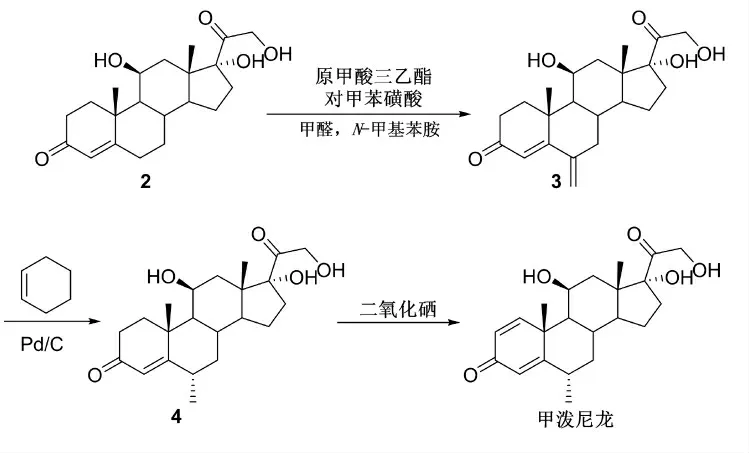
Scheme 1
化合物6α-甲基氢化产物(1)是合成甲泼尼龙的关键中间体,韩朝东[5]和刘东华[6]等以6α-甲基氢化产物为起始原料,经过生物发酵、卤代、亲核取代、水解等步骤可制备得到甲泼尼龙(Scheme 2)。

Scheme 2
汪家振[7]公开了一种甲基泼尼松龙中间体6α-甲基氢化产物的化学合成方法(Scheme 3),以霉菌氧化物为起始原料,依次经普氏氧化、溴代、脱溴、6 位次甲基化、6 位甲基化、缩酮保护、还原、水解等8 步反应可制备得到甲基泼尼松龙中间体6α-甲基氢化产物(1)。该反应路线较长,需要用到三氧化铬和氯化锰等重金属氧化剂等,产生的废水污染较大,不适合工业生产,同时脱溴反应需要加压条件下的氢气和金属镍。
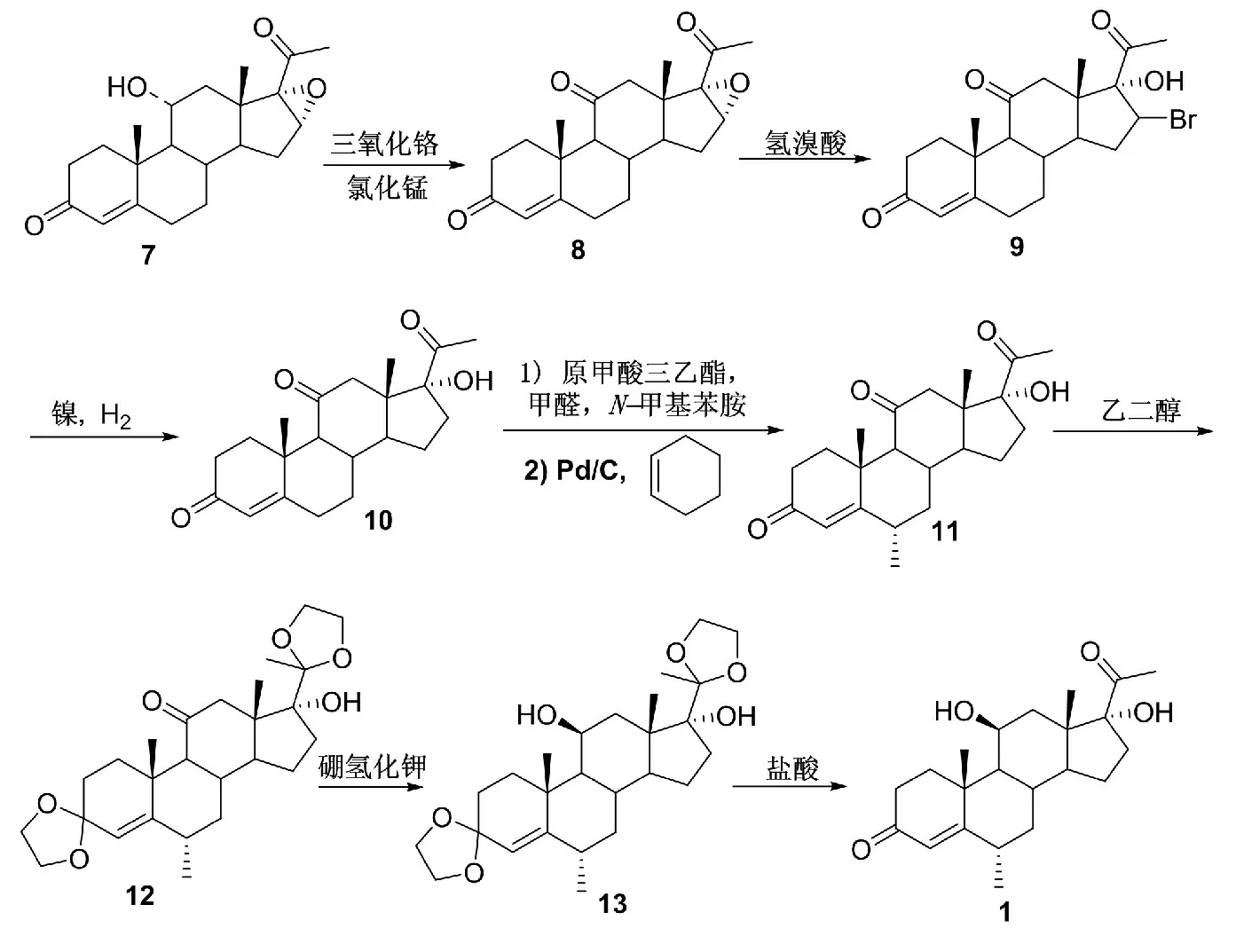
Scheme 3
Natsuko 等[8]以化合物14 为起始原料,经酯化反应、缩酮保护、环氧化、格氏反应、水解、消除等6 步反应以23%的收率制备得到甲基泼尼松龙中间体6α-甲基氢化产物(1)(Scheme 4)。
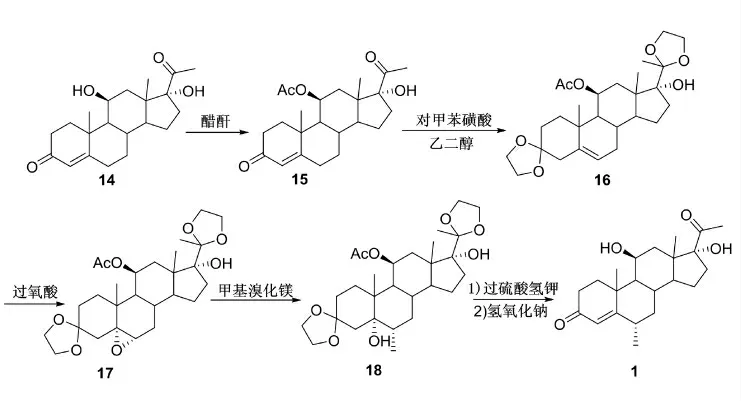
Scheme 4
本研究在文献[8]的路线基础上作进一步改进(Scheme 5),主要改进点有以下两方面:(1)整体路线以54%的收率制备得到甲基泼尼松龙中间体6α-甲基氢化产物(1),相比之前的工艺路线收率提高了1 倍以上;(2)格氏反应、缩酮脱保护、消除3 个步骤中间产物无需分离,直接制备得到6α-甲基氢化产物(1),其收率达65%以上。不仅简化了制备步骤,降低了工艺成本,同时提高了总收率。

Scheme 5
1 实验部分
1.1 主要原料
化合物10 为企业自产;其余原料及试剂均为试剂级,从安耐吉、阿拉丁等试剂有限公司购得。
1.2 化合物的合成
1.2.1 19 的合成
将乙二醇(7.0 g)、原甲酸三乙酯(21 mmol,3.2 g)、化合物10(8.7 mmol,3.0 g)及50 mL 二氯甲烷加入200 mL 圆底烧瓶中,室温下搅拌5 min。加入对甲苯磺酸(0.87 mmol,0.15 g),于室温下继续反应6~8 h,薄层层析(TLC)监测反应进度。反应完毕,加入三乙胺调节反应液pH=7~8,水洗、分液得有机层,真空下减压浓缩得化合物19。
1.2.2 20 的合成
向100 mL 圆底烧瓶中加入上述所得化合物19 及36 mL 四氢呋喃。缓慢加入1.5 g 硼氢化钠,加料完毕。升温至60 ℃,保温24 h,TLC 监测反应进度。待反应结束,真空减压浓缩,加冰醋酸调pH=6~7,向反应瓶中加入水并搅拌,可观察到有固体析出,搅拌1 h,过滤,烘干得还原产物20,收率为98%。
将上述还原产物20 加入50 mL 圆底烧瓶中,加入20 mL 氯仿溶解完全,降温至0 ℃~5 ℃,搅拌30 min,滴加间氯过氧苯甲酸,控制滴加温度为0 ℃左右。滴加完毕,于30 ℃保温反应8 h以上。反应结束后,用三乙胺调节反应液pH=8,加入水萃取,得有机层,用无水硫酸钠干燥,减压浓缩至干,得化合物20。收率85%。
1.2.3 1 的合成
将化合物20 加入三口圆底烧瓶中,加入四氢呋喃,通氮气保护,搅拌,开始缓慢加入甲基氯化镁,控制温度不高于50 ℃。格氏试剂加完后,升温回流反应4~5 h。
在圆底烧瓶中加入2 mL 冰醋酸、12 mL 水,降温至0 ℃,将上述格氏反应液缓慢加入酸水中,控制温度为50 ℃以下。加入二氯甲烷搅拌萃取,静置分层,分离得有机层。
向上述有机层中加入硫酸水溶液,体系逐渐析出白色固体,于室温下保温反应2 h,缓慢滴加NaOH 溶液,取样TLC 跟踪反应完全后,降温至50 ℃以下,加冰醋酸调节pH=6~7,pH 调节完毕,减压浓缩,水析,过滤得固体。用甲醇重结晶得化合物1,收率为65%。
1H NMR (400 MHz,CDCl3) δ 5.74 (s,1H),4.52~4.44 (m,1H),2.95~2.66 (m,2H),2.61~2.33(m,4H),2.29 (s,3H),2.21~1.83 (m,7H),1.77~1.47(m,5H),1.44 (s,3H),1.10~1.01 (m,6H)。13C NMR(100 MHz,CDCl3) δ 211.4,199.7,175.3,119.7,89.5,68.6,55.9,51.4,47.7,41.9,39.8,39.5,35.1,33.5,33.4,33.2,31.1,27.8,23.9,22.2,18.2,18.0。
2 结果与讨论
2.1 环氧化物的合成
控制单一变量,考察了m-CPBA 的用量和反应温度对环氧化物收率的影响。随着m-CPBA 的用量不断增加,收率逐渐提高,当m-CPBA 的摩尔比大于1.5 时,环氧化物的产率没有明显增加,且过量的过氧酸可能会引发副产物的生成,收率反而降低(图2)。
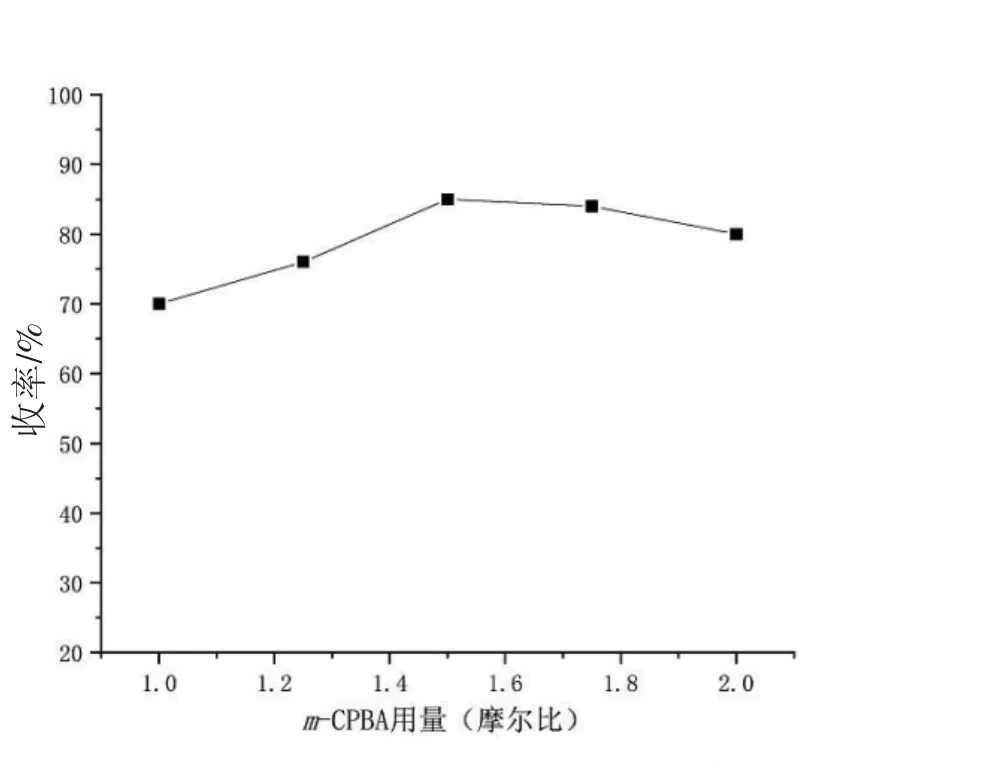
图2 m-CPBA 用量过反应收率的影响
温度对反应收率的影响见图3。分别设置了0 ℃、10 ℃、20 ℃、30 ℃、40 ℃等5 个梯度。从图3中也看出,随着温度的提高,产率不断提高;但当反应升温度大于等于30 ℃时,收率不再明显上升。综上所述,得出环氧化产物的最优条件为以二氯甲烷或者三氯甲烷为溶剂,m-CPBA(间氯过氧苯甲酸)摩尔比为1.5,在30 ℃下反应8 h。
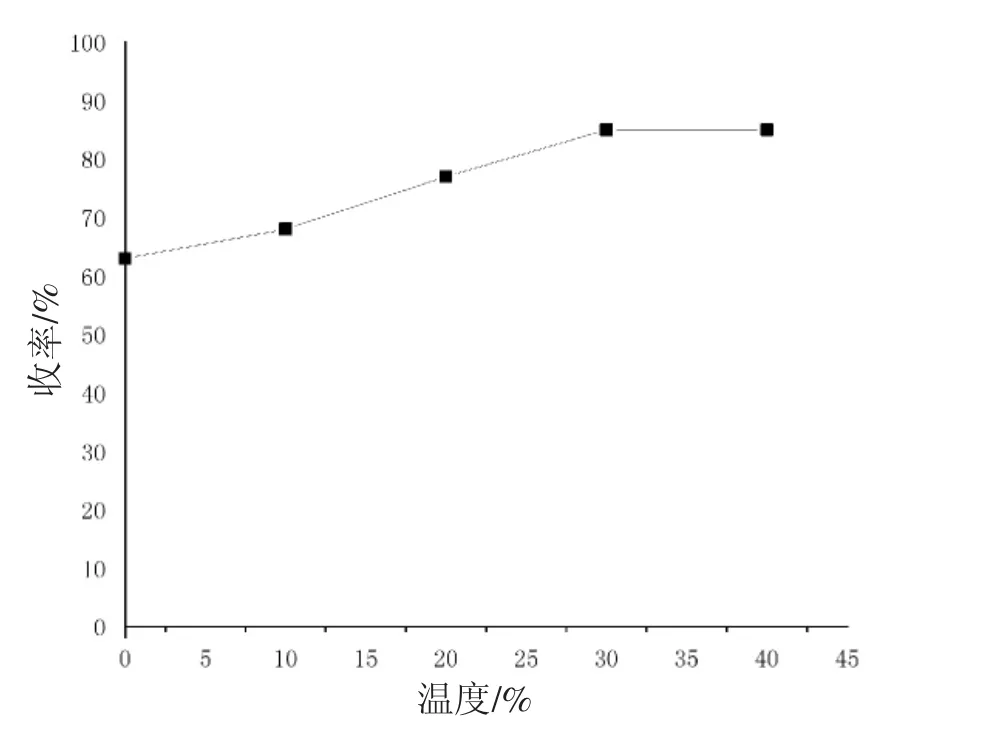
图3 温度对反应收率的影响
2.2 格氏反应的条件优化
化合物20 经过与格氏试剂的环氧开环在C6位引入甲基。考察了格氏试剂(CH3MgCl)用量,反应温度及反应溶剂对反应收率的影响,结果见表1。在控制反应温度相同的条件下,减少或增加甲基氯化镁的用量均无法促进化合物20 收率的提高(摩尔比1.5 为较优用量)。随后,对反应温度作进一步筛选,结果表明65 ℃是较优的反应温度,收率可达82%。

表1 格氏反应影响因素研究Tab.1 Screening of Grignard reaction
2.3 中间体21 不分离条件探索
中间体21 经格氏反应、脱保护及消除等3步反应可制备得到化合物1,传统方法中间体21需通过结晶分离。在跟踪反应过程中,发现格氏反应的选择性较好,因此尝试了中间体21 不经过分离,直接投入后续脱保护及消除反应,以期能减少分离步骤,提高反应总收率。结果表明,若中间体21 不分离,格氏反应、脱保护及消除三步总收率可达到65%;若通过结晶方法分离中间体21,则三步反应总收率仅为58%。
3 结论
综上所述,以醋酸可的松氧化中间体17α-羟基孕甾-4-烯-3,11,20-三酮作为原料,经过缩酮保护、硼氢化钠还原、m-CPBA 环氧化、格氏反应、水解、消除等反应得到甲泼尼龙关键中间体6α-甲基氢化产物(1)。其中,格氏反应、脱保护水解、消除等3 个步骤得到的中间体无需分离,直接制备得到甲泼尼龙关键中间体6α-甲基氢化产物(1),其收率达65%以上,最终总收率达54%。该方法简化了操作步骤,降低了工艺成本,同时提升了反应总收率。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
纺织科学研究(2020年1期)2020-05-21 00:31:06
小学科学(学生版)(2019年7期)2019-08-01 09:57:52
现代塑料加工应用(2016年6期)2016-02-28 17:47:53
科学启蒙(2015年9期)2015-09-25 03:56:58
橡胶工业(2015年2期)2015-07-29 08:29:46
西南军医(2014年5期)2014-04-25 07:42:49
食品工业科技(2014年9期)2014-03-11 18:15:39
无机化学学报(2014年12期)2014-02-28 17:34:01
