
Charcot-Marie-Tooth-1A and sciatic nerve crush rat models: insights from proteomics
2023-01-21 04:42ZeinaMsheikStephanieDurandEmiliePinaultMartialCaillaudLaetitiaVignaudFabriceBilletMohamedElMassryAlexisDesmouliere
中国神经再生研究(英文版) 2023年6期
Zeina Msheik,Stephanie Durand,Emilie Pinault,Martial Caillaud,Laetitia Vignaud,Fabrice Billet,Mohamed El Massry,Alexis Desmouliere,
Abstract The sensorimotor and histological aspects of peripheral neuropathies were already studied by our team in two rat models: the sciatic nerve crush and the Charcot-Marie-Tooth-1A disease.In this study,we sought to highlight and compare the protein signature of these two pathological situations.Indeed,the identification of protein profiles in diseases can play an important role in the development of pharmacological targets.In fact,Charcot-Marie-Tooth-1A rats develop motor impairments that are more severe in the hind limbs.Therefore,for the first time,protein expression in sciatic nerve of Charcot-Marie-Tooth-1A rats was examined.First,distal sciatic nerves were collected from Charcot-Marie-Tooth-1A and uninjured wild-type rats aged 3 months.After protein extraction,sequential window acquisition of all theoretical fragment ion spectra liquid chromatography and mass spectrometry was employed.445 proteins mapped to Swiss-Prot or trEMBL Uniprot databases were identified and quantified.Of these,153 proteins showed statistically significant differences between Charcot-Marie-Tooth-1A and wild-type groups.The majority of these proteins were overexpressed in Charcot-Marie-Tooth-1A.Hierarchical clustering and functional enrichment using Gene Ontology were used to group these proteins based on their biological effects concerning Charcot-Marie-Tooth-1A pathophysiology.Second,proteomic characterization of wild-type rats subjected to sciatic nerve crush was performed sequential window acquisition of all theoretical fragment ion spectra liquid chromatography and mass spectrometry.One month after injury,distal sciatic nerves were collected and analyzed as described above.Out of 459 identified proteins,92 showed significant differences between sciatic nerve crush and the uninjured wild-type rats used in the first study.The results suggest that young adult Charcot-Marie-Tooth-1A rats (3 months old) develop compensatory mechanisms at the level of redox balance,protein folding,myelination,and axonogenesis.These mechanisms seem insufficient to hurdle the progress of the disease.Notably,response to oxidative stress appears to be a significant feature of Charcot-Marie-Tooth-1A,potentially playing a role in the pathological process.In contrast to the first experiment,the majority of the proteins that differed from wild-type were downregulated in the sciatic nerve crush group.Functional enrichment suggested that neurogenesis,response to axon injury,and oxidative stress were important biological processes.Protein analysis revealed an imperfect repair at this time point after injury and identified several distinguishable proteins.In conclusion,we suggest that peripheral neuropathies,whether of a genetic or traumatic cause,share some common pathological pathways.This study may provide directions for better characterization of these models and/or identifying new specific therapeutic targets.
Key Words: Charcot-Marie-Tooth-1A;endoplasmic reticulum;Gene Ontology;neurogenesis;oxidative stress;proteomics;rat;repair;sciatic nerve crush;SWATH-MS
From the Contents
Introduction 1354
Methods 1355
Results 1356
Discussion 1361
Introduction
Peripheral neuropathy (PN) is one of the most prevalent neurological conditions in humans (Watson and Dyck,2015).It manifests with varying severity (Martyn and Hughes,1997) and a panoply of symptoms,ranging from small sensory defects to the more complex motor and autonomic symptoms(Barrell and Smith,2019).Etiologically,PN can arise due to either traumatic damage to the nerve,or non-traumatic damage as seen in genetic,metabolic,drug-induced,and immune-related forms.Charcot-Marie-Tooth (CMT) is the most common inherited form of hereditary neuropathy with a worldwide prevalence of 1:2500 (Barreto et al.,2016),with demyelinating type 1A being the most abundant subtype (Barreto et al.,2016).Whether PN is of genetic origin or not,it has a significant impact on the quality of life of patients.Importantly,effective therapies for neuropathies are limited,especially due to patient heterogeneity and incomplete understanding of the underlying pathological mechanisms (Morena et al.,2019).
To further study the mechanistic alterations in PN and to screen for potential targets for drug design,animal models were developed for both the genetic and non-genetic forms of PN.For CMT1A disease,a transgenic rat model harboring three additional murine copies of PMP22 gene,fairly reproduces the sensorimotor symptoms observed in human patients (Sereda et al.,1996;Fledrich et al.,2012).Furthermore,sciatic nerve crush (SNC) or axonotmesis is one of the most common models of peripheral nerve injury in rodents(Magill et al.,2007).Crushing interrupts all axons,while Schwann cell basal laminae are preserved so that recovery is possible (Amado et al.,2008;Luís et al.,2008).
The functional characterization of disease-associated proteins is a major challenge in the post-genomic era.Proteins are effectors of biological function,and their levels not only depend on corresponding mRNA levels but also on host translational regulation.In the last 10 years,mass spectrometrybased proteomic analysis has revolutionized the way proteins can be analyzed and detected in fluids,cells,and tissues (Meissner and Mann,2014).Therefore,analysis of the nerve proteome could provide useful information for the characterization of the pathophysiology of PN and the identification of potential biomarkers or targets for therapy.
The present proteomic study is a follow-up work of previously conducted experiments (Caillaud et al.,2018,2020),and was performed using distal sciatic nerve samples extracted from three groups of animals: CMT1A rats,SNC rats,and wild-type (WT) uninjured rats.Sequential window acquisition of all theoretical fragment ion spectra mass spectrometry (SWATH-MS) is an emerging technology that combines deep proteome coverage capabilities with quantitative consistency and accuracy (Ludwig et al.,2018).We aimed to explore variations in protein expression of the CMT1A rat model and protein variations 1 month after SNC injury.
Methods
Animals and proteomic study organization
The workflow of the study is summarized inFigure 1.
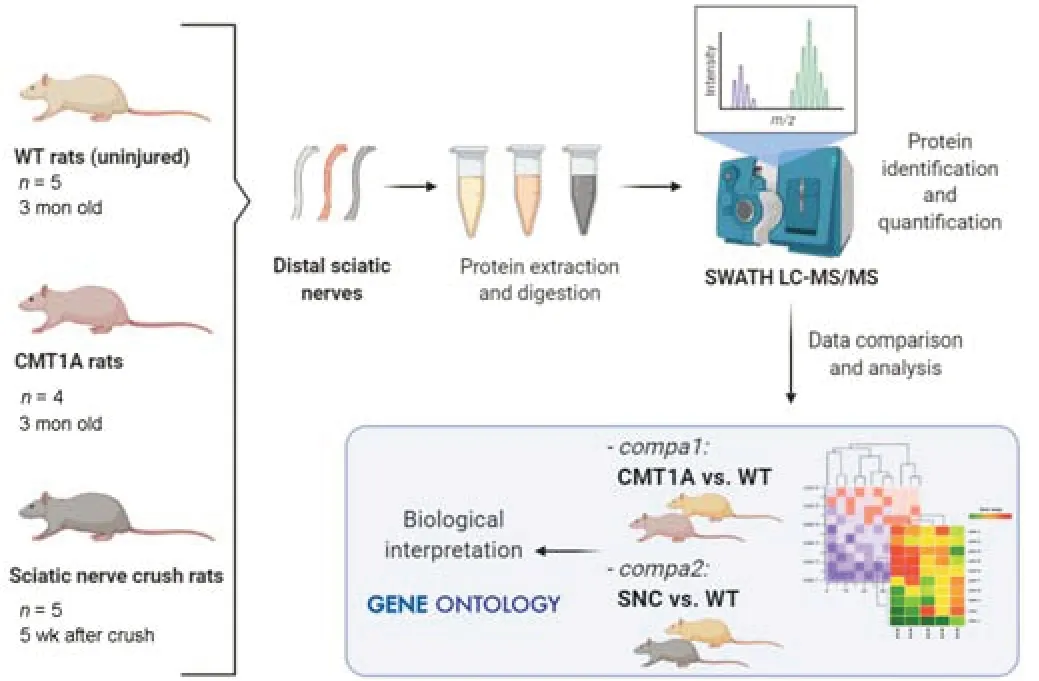
Figure 1|Experimental workflow.
Sciatic nerves from wild-type (WT) male Sprague-Dawley rats (Janvier Labs,Le Genest-Saint-Isle,France) aged 3 months (n=5) and of body weight of 250–300 g;and age-and weight-matched male CMT1A heterozygous rats(generated on a Sprague-Dawley background (Sereda et al.,1996) by the Max Planck Institute for Experimental Medicine (MPIEM,University of Göttingen,Göttingen,Germany);n=4) were collected post mortem.The CMT1A rats harbor 3 copies of murinePmp22gene and present motor and sensory symptoms similar to those observed in human patients (Sereda et al.,1996).Genotyping of male CMT1A rats was performed by quantitative polymerase chain reaction (qPCR) analysis of DNA samples.Genomic DNA was purified from tail biopsies using the DNeasy blood and tissue kit (Qiagen,Hilden,Germany) according to the manufacturer’s instructions.The genotype of either CMT1A rats or WT rats was confirmed by the presence or absence of Pmp22 transgenes detected by using mouse-specificPmp22primers (forward primer: GTT CCT GTT CTT CTG CCA GC and reverse primer CCT CAT TCG CGT TTC CGC A) (Thermo Fisher Scientific,Waltham,MA,USA).
For the traumatic model,SNC was performed on male WT Sprague-Dawley rats aged 1 month (n=5) as previously described in detail (Caillaud et al.,2018).Briefly,after inhalation anesthesia by isofluorane (ISO-VET,Voorschoten,Netherlands),unilateral SNC was applied via a non-serrated clamp exerting a constant force using a Stevens’s needle holder (World Precision Instruments,Berlin,Germany) clamped directly onto the sciatic nerve 1.5 cm above the bifurcation,for 30 seconds to create a crush injury 2 mm in length.The jaws of the clamp were coated with carbon powder to precisely delimit the crush site.Muscles and skin were sutured,and a subcutaneous injection of analgesic (buprenorphine 0.05 mg/kg,Axience SAS,Pantin,France) was carried out after surgery,once a day for 3 days.Just after the crush injury,rats showed a deterioration in sensorimotor function,which corresponds to partial nerve damage and subsequent degeneration.Animals were then allowed to recover for 5 weeks.During this period,and at 5 weeks,the rats showed improvements at the functional and histological levels,corresponding to partial repair (see the behavioral study in Caillaud et al.,2018).Sciatic nerves were collected post-mortem (age at sacrifice 9 weeks).
Animals were individually housed in cages with an enriched environment and maintained on a 12-hour light/dark cycle,at a room temperature of 22°C,with ad libitum access to food and water.All animal experimentation was performed according to recommendations of the European Directive on 22 September 2010 (2010/63/EU) for the protection of animals used for scientific purposes.SNC experimental protocol and CMT1A animal model maintenance were specifically approved by the ethical committee CREEAL (Regional Animal Experimentation Ethics Committee) (n°16-2013-16 and APAFIS#29437-2021020115213118v1).Animal studies are reported in compliance with the ARRIVE 2.0 guidelines (Animal Research: Reporting ofIn VivoExperiments)(Percie du Sert et al.,2020).Every effort was made to minimize the number of animals used and to ensure their optimal well-being before,during,and after each experiment.All rats were observed daily for general well-being. Note that the data from the uninjured WT rat group (aged 3 months) served as a control for both experiments.
Proteomic profiles were compared between CMT1A and WT rats sacrificed at 3 months old (compa1) and between SNC rats sacrificed 5 weeks after crush injury (compa2).In addition,proteins from a separate group of rats,both male and female,were examined by western blotting (see below).
Extraction and preparation of sciatic nerve protein samples
Nerve tissue samples were placed in liquid nitrogen and ground to a fine powder with mortar and pestle,suspended in 10 volumes of lysis buffer (8 M urea,2 M thiourea,4% w/v CHAPS,40 mM Tris,50 mM DTT,1 mM EDTA and protease inhibitor cocktail),vortexed,sonicated at 4°C and allowed to sit on ice for 2 hours.Crude extracts were centrifuged at 17,000 ×gfor 15 minutes and supernatants were collected.Protein concentration was determined using PlusOne 2D Quant Kit (GE Healthcare Bio-Sciences AB,Schnelldorf,Germany)as per kit instructions.
Protein digests
Aliquots of 50 μg were prepared from supernatants by filter aided sample preparation (FASP) method as follows: protein extracts were diluted in 8 M urea to a final concentration of 0.5 μg/μL,reduced with 0.1 volume of 50 mM DTT and alkylated with 0.1 volume of 100 mM iodoacetamide.The protein extract was then transferred into the Amicon Ultra 10K filter (Millipore,Darmstadt,Germany) and washed twice with 8 M urea and then twice with 25 mM ammonium bicarbonate.Trypsin was added at a 1:50 ratio for a final concentration of 10 ng/μL.Peptides were purified using a 1 mL 30 mg Hydrophilic-Lipophilic-Balanced (HLB) cartridge following the manufacturer’s protocols (Waters,Milford,MA,USA) and finally filtered using 0.22 μm spin columns (Agilent Technologies,Santa Clara,CA,USA).
Mass spectrometry
Peptides were analyzed by micro LC-MS/MS using a nanoLC 425 system in micro-flow mode (Eksigent,Dublin,CA,USA) coupled with time-of-flight (TOF)(TripleTOF 5600+Sciex,Framingham,MA,USA) operating in high-sensitivity mode.Reverse-phase LC was performed via a trap and elute configuration using a trap column (C18 Pepmap100 cartridge,5 μm pore size;Thermo Fisher Scientific) and an analytical column (ChromXP C18 column,12 nm,3 μm pore size,Sciex) with the following mobile phases: loading solvent (water/ACN/TFA 98/2/0.05% (v/v)),solvent A (0.1% (v/v) TFA in water) and solvent B (water/ACN/TFA 5/95/0.1% (v/v)).All samples were loaded,trapped,and desalted using a flow rate of 10 μL/min with loading solvent for 5 minutes.The chromatographic separation was performed at a flow rate of 2 μL/min as follows: initial,5% solvent B,increased to 25% for 90 minutes,then increased to 95% B for 10 minutes,maintained at 95% for 5 minutes and,finally,decreased to 5% B for re-equilibration.
The SWATH-MS spectral library was generated from 1 μg of each sample by data-dependent acquisition.MS and MS/MS data were continuously recorded with up to 40 precursors selected for fragmentation from each MS survey scan.Precursor selection was based upon ion intensity and whether or not the precursor had been previously selected for fragmentation (dynamic exclusion).Ions were fragmented using rolling collision energy.One μg of each sample was then subjected to cyclic data-independent acquisition of mass spectra using 60 variable SWATH windows over the 400–1250 m/z range.A 50 ms survey scan was initially performed on a TOF mass spectrometer,and subsequent MS/MS experiments were performed on all precursors using an accumulation time of 75 ms per SWATH window,for a total cycle time of 4.6 seconds.Ions were fragmented using rolling collision energy corresponding to the window m/z range.
Generation of the SWATH-MS reference library
All data-dependent acquisition mass spectrometer files were searched using ProteinPilot software v.5.0 (Sciex) with the Paragon algorithm.Samples were input with the following parameters: cysteine alkylation with iodoacetamide,digestion by trypsin,and no special factors.The search was conducted using a rapid identification effort of a UniProt https://www.uniprot.org/ (SWISSPROT or TrEMBL) database (February 2018 release) containing non-redundant proteins ofRattus norvegicus.Group file output of this search was used as the reference spectral library.Spectral alignment and targeted data extraction of data-independent acquisition samples were performed using PeakView v.2.1(Sciex) and SWATH v.2.0 module with the reference spectral library as above.Fold change (FC) of each protein was calculated as FC (protein)=mean of tested groups/mean of WT groups.
Principal component analysis and hierarchical clustering
To evaluate differences among samples,multivariate analysis was performed using principal component analysis (PCA) with FactoMineR (Lê et al.,2008) and visualized using factoextra (Kassambara and Mundt,2020).A heat map is a useful tool for presenting quantitative proteomic datasets organized as matrices.Hierarchical clustering analysis was performed after log2 transformation and median centering,using ComplexHeatmap,according to Pearson distance to calculate dissimilarity and the average method to agglomerate samples that showed similar protein expression profiles (Gu et al.,2016).
Protein-protein interaction network and Functional enrichment analysis
The search tool for the retrieval of interacting genes (STRING) (https://string-db.org/cgi/input.pl) is an online tool designed to evaluate the proteinprotein interaction (PPI) network (Szklarczyk et al.,2015).Full PPI networks of CMT1Avs.WT (compa1) and SNCvs.WT (compa2) were constructed by STRING,and no maximum number of interactors was set.The cut-off criterionP< 0.05 was set for pathways enrichment analysis.Markov Clustering (MCL)of nodes was applied.Gene ontology (GO) (Gene Ontology Resource;http://geneontology.org/) and Pathway ((Kyoto encyclopedia of genes and genomes(KEGG) (KEGG PATHWAY Database;https://www.genome.jp/kegg/pathway.html) and Reactome (Home-Reactome Pathway Database;https://reactome.org/) databases)) enrichment analyses were performed using the database for annotation,visualization and integrated discovery (DAVID®),v.6.8 (https://david.ncifcrf.gov/) (Huang et al.,2009) which provides comprehensive functional annotations to understand biological meaning behind large lists of genes.The GO describes our knowledge of the biological domain with respect to three aspects: molecular function (MF),biological process (BP),and cellular component (CC).Entities like nucleic acids,proteins,and small molecules participate in reactions to form a network of biological interactions and are grouped into pathways.The results were filtered by the following criteria: p-adjusted (Benjamini-Hochberg correction) ≤ 0.05 and protein count ≥ 5.For a given GO term,fold enrichment is defined as the ratio of the two proportions: proportion of input genes associated with the GO term/proportion of background genes (all genes ofRattus norvegicus) associated with the GO term.
Western blotting
Western blotting was used to verify results for expression of certain proteins seen by proteomic analysis for compa1.Sciatic nerves were obtained from 3-month-old CMT1A (n=8: 4 males and 4 females) and littermate WT rats (n=7: 4 males and 3 females).These rats have the same general information as the above-mentioned rats.Frozen nerves were crushed on dry ice and solubilized in PierceTMRIPA lysis buffer (ThermoFisher Scientific) containing protease inhibitor cocktail (cOmplete Mini Tablets,EDTA-free,Roche,Switzerland).The samples were homogenized using a Tissuelyser (Qiagen,Hilden,Germany) sonicated on ice and centrifuged for 10 min at 14,000 ×g.Protein concentrations from cell lysates were quantified using Bradford solution (Sigma-Aldrich,St.Louis,MO,USA).Twenty-five μg of total protein was separated on a 4–20% polyacrylamide mini-gel (Biorad,Hercules,CA,USA),and transferred to a PVDF membrane.Gels were stained by Coomassie blue and blots blocked with 5% bovine serum albumin (BSA) in TBST.
All antibody incubations were overnight at 4°C.Antibodies used were anticalnexin mouse monoclonal antibody (1:10,000,Proteintech,Cat# 66903-1-Ig,RRID: AB_2882231),anti-PMP22 rabbit polyclonal antibody (1:200,Sigma-Aldrich,Cat# SAB4502217,RRID: AB_10746275),anti-peripherin rabbit polyclonal antibody (1:1000,Santa Cruz Biotechnology,Cat# sc-28539,RRID:AB_2171206),anti-vimentin mouse monoclonal antibody (1:75,Agilent,Cat# M0725,RRID: AB_10013485),anti-aldose reductase ALDR (H-6) mouse monoclonal antibody (1:500,Santa Cruz Biotechnology,Cat# sc-166918,RRID: AB_10609906),or anti-heat shock protein GRP94 rabbit polyclonal antibody (1:1000,Enzo Life Sciences,Cat# ADI-SPA-851,RRID: AB_10615790).Following incubation,membranes were washed and probed for 1h at room temperature with anti-rabbit or anti-mouse secondary antibodies conjugated to horseradish peroxidase (HRP) (1:1000,Rabbit: Agilent,Cat# P0448,RRID:AB_2617138,Mouse: Agilent,Cat# P0447,RRID: AB_2617137).
Protein band intensities were quantified using ImageJ software v1.53c(National Institutes of Health,Bethesda,MD,USA).Protein quantification was calculated as follows: intensity of the protein of interest/intensity of total proteins per lane (in arbitrary units).
Statistical analyses
For the proteomic study,statistical analyses were performed using R environment (version 3.6.2) (R: The R Project for Statistical Computing,Vienna,Austria;https://www.r-project.org/).Normality and homoscedasticity of protein levels data according to treatment group were estimated by Shapiro and Bartlett tests,respectively.Where non-normal distribution,heteroscedasticity,and small group size (≤ 5) occurred,subsequent analysis by non-parametric tests was carried out to identify differentially expressed proteins between different groups.Two-group comparisons: CMT1Avs.WT(compa1) and SNCvs.WT (compa2) by Mann-WhitneyUtest,withP-value <0.05 considered statistically significant.To control for multiple testing of the average expression of proteins,the rawP-values obtained from this analysis were further adjusted with the Benjamini-Hochberg method.In SNCvs.WT study (compa2),we were obliged to use a non-adjustedP-value,as the P-adjusted was not significant,potentially due to greater heterogeneity of individuals.Significance was set atP< 0.05.
For the western blot study,statistical analyses were performed using GraphPad Prism version 8.3.0 for Windows (GraphPad Software,San Diego,CA,USA,www.graphpad.com).The Student’st-test was used,and statistical significance was considered whenP< 0.05.
Results
Protein detection by the SWATH LC-MS/MS strategy allowed the identification of 445 proteins from CMT1A samples and 459 proteins from SNC samples.Each disease model group (CMT1A or SNC) was compared to the WT group.Dysregulated genes were filtered by the level of significance (Pvalue) and magnitude of the regulation (fold change).
CMT1A versus WT (compa1)
Between CMT1A and WT groups,153 proteins showed statistically-significant differential expression (P< 0.05).A first visual exploration of the proteomic data was conducted via a 2D PCA plot of the 153 significantly different proteins (Figure 2A).As expected,the two groups were well separated.WT samples were closely clustered,suggesting a similarity in protein expression between samples of this group.In contrast,CMT1A samples were more widespread,forming a larger distribution area and suggesting inter-individual heterogeneity among CMT1A samples.
For each protein,fold change (FC) ratios (mean CMT1A/mean WT) greater than 2 or less than 0.5 were designated to consider a protein over or underexpressed,respectively.The majority of affected proteins were overexpressed in CMT1A in comparison to the WT group,with 99 proteins upregulated in CMT1A (FC > 2),whereas only 7 proteins were downregulated in CMT1A (FC< 0.5) as compared to WT rats (Figure 2B).These results were visualized using the heat map (Figure 3) that orders data with similar profiles into cluster rows and columns so that patterns among the data can be more easily observed.
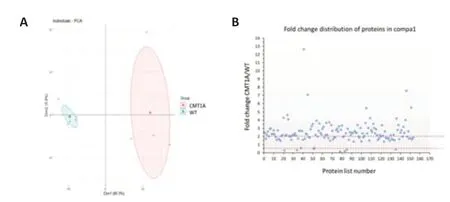
Figure 2|Data visualization of proteins sorting from compa1 (153 proteins).
STRING analysis was employed to check for protein-protein interactions (PPI)of the 153 significantly different proteins found in compa1 (Figure 4).As many proteins have more than one function,STRING bioinformatics tool was used to get a more detailed,yet simplified view of PPI.As expected,this network demonstrated significantly more interactions than would be expected among a random set of proteins (PPI enrichmentP-value < 0.001).Using clustering techniques,similar proteins were indicated by the same color.Each colored node represents a protein,i.e.all the transcripts produced by a single,protein-coding gene locus.Therefore,splice isoforms or post-translational modifications were collapsed,resulting in a final 143 nodes.
To facilitate the analysis of such a large data set,differentially expressed proteins were categorized using Gene Ontology Biological Process (GO-BP)terms and functional enrichment analysis performed with DAVID® software.Ninety-eight GO-BP terms were identified as enriched (P< 0.05).These terms were manually grouped into 14 “families” based on similarity (Figure 5).Some of the proteins were involved in multiple processes and were therefore assigned to more than one BP group.Regarding CMT1A physiopathology,several BP terms that emerged from this analysis would be relevant to the disease process,including neurogenesis,gliogenesis,protein folding,cell projection and cytoskeleton organization,metabolic processes,wound healing,and regeneration.Although not the largest in terms of the number of proteins,we also noted with particular interest the category of response to oxidative stress.
One day, while the girl was on her way to work, she was knocked down by a car that lost control. When she woke up, she saw her parents beside her bed. She realized that she was badly injured. Seeing her mum crying, she wanted to comfort her but to find that all that could come out of her mouth was just a sigh. She had lost her voice ...
To determine the most pronounced BP terms,we refined the data based on statistical significance and fold enrichment.Data were filtered byP-value,and a further filter of fold enrichment > 5 was applied (Additional Table 1).The top-five terms were selected (Table 1).Interestingly,the top 2 BP terms,containing 14 and 10 proteins respectively,are related to the response to reactive oxygen species (ROS).Arrangement in a Venn diagram demonstrated that 17 irredundant proteins from the 4 terms related to oxidative stress were determined (Figure 6A).The protein list contains thioredoxin (R4GNK3),peroxiredoxin-1,2,and 5 (PRDX,Q63716,P35704,and Q9R063,respectively),superoxide dismutase [Cu-Zn] (SOD1,P07632),macrophage migration inhibitory factor (MIF,P30904),glutathione S-transferase P (GST-p,P04906),heat shock cognate 71 KDa protein (HSC70,P63018),and aldo-keto reductase family 1 member B1 (P07943,ALDR).
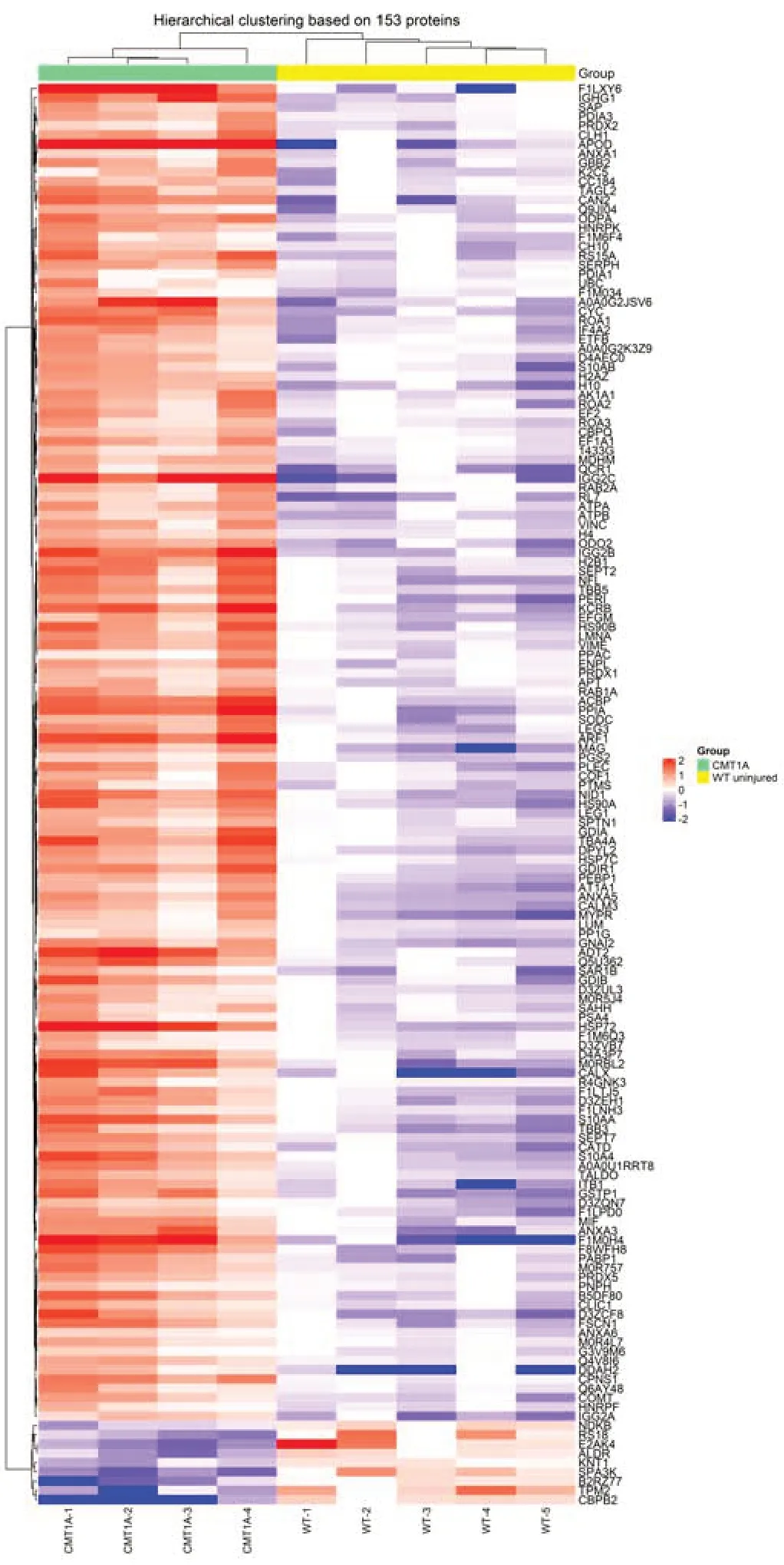
Figure 3|Heat map of CMT1A versus WT (compa1).
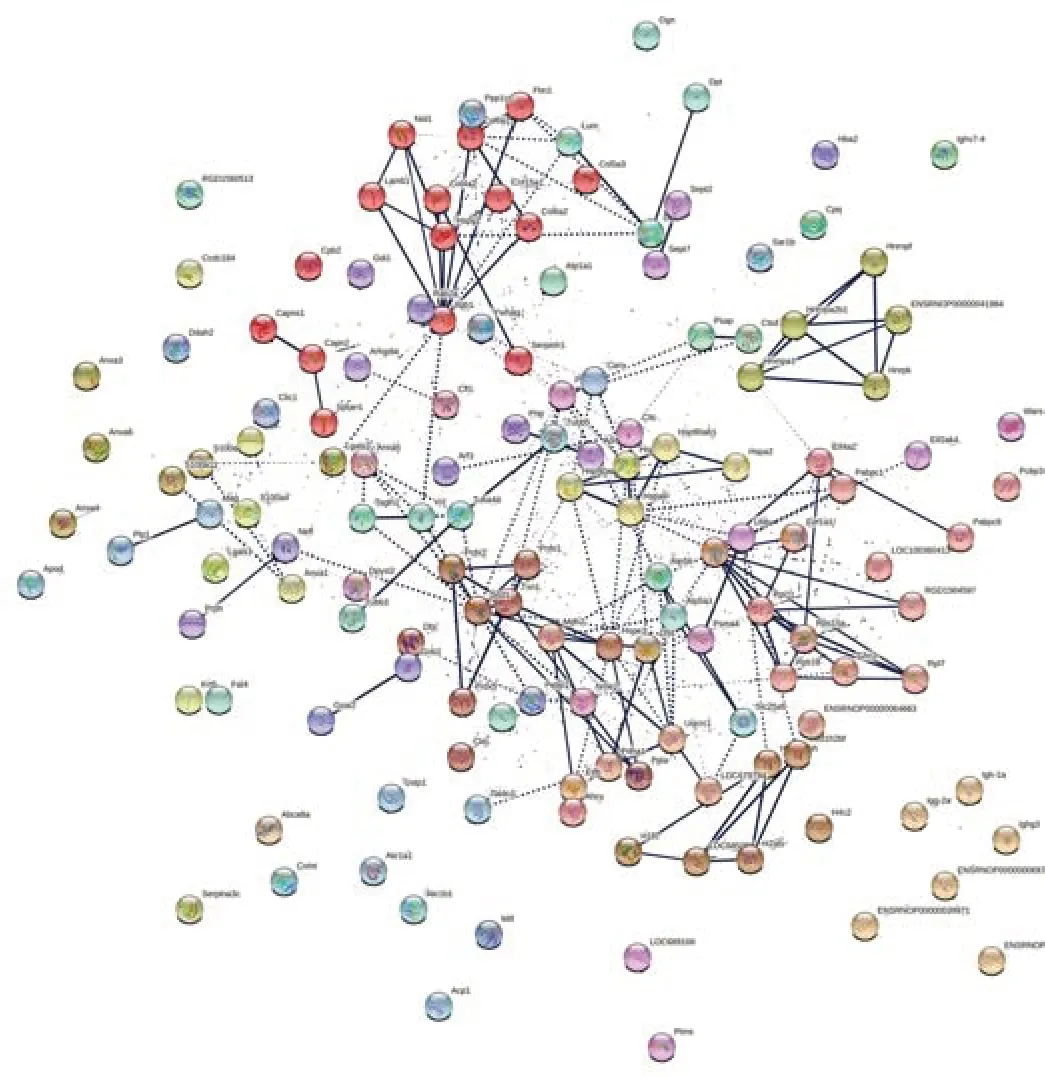
Figure 4|Protein network data of compa1 by STRING database (Rattus norvegicus).
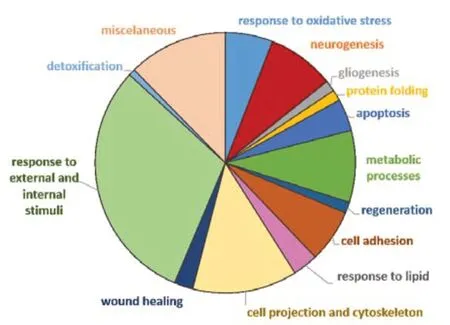
Figure 5|Pie chart of dysregulated proteins in CMT1A versus WT (compa1)annotated to Gene Ontology-Biological Process (GO-BP) terms.
Moreover,third in the top-five list comes the term “protein folding” with 7.38 fold enrichment (Table 1).This group contains several heat shock proteins that are designated by their molecular weight: heat shock cognate 71 kDa protein (HSC70,P63018),heat shock protein 90-alpha (HSP90-a,P82995),and beta (HSP90-b,P34058),heat shock-related 70 kDa protein 2 (HSP70.2,P14659),and the 10 kDa mitochondrial heat shock protein(HSP10,P26772).In addition,we found other molecular chaperones,most of which are responsible for glycoprotein folding such as endoplasmin (GRP94,Q66HD0),calnexin (CNX,P35565),peptidyl-prolyl cis-trans isomerase A(PPIase A,P10111),protein disulfide-isomerase A3 (PDIA3 or ERp57,P11598),and thioredoxin (TRX,R4GNK3).In CMT1A samples,these proteins were significantly upregulated.Our data show that CNX,GRP94,proteasome subunit alpha type-4 (PSA4,P21670),and polyubiquitin-C (UBC,Q63429)were upregulated in CMT1A samples with FC=4.246,2.208,1.604 and 1.637,respectively.These results suggest the activation of the endoplasmic reticulum-associated degradation (ERAD) pathway which eliminates terminally misfolded glycoproteins (PMP22 in this case).Indeed,this pathway involves CNX,GRP94,the proteasome,and the ubiquitin system (Volpi et al.,2017).However,the ER chaperone BiP (also known as GRP78,P06761),a direct ER stress sensor (Wang et al.,2017),remained unchanged in CMT1A in comparison to WT.
We sought to investigate a potential interaction between the 17 proteins related to oxidative stress and the 10 proteins related to protein folding that came out as significantly changed in CMT1A investigated via STRING.The interactions (PPI) of these proteins were shown inFigure 6B.Significantly,this network showed a high level of interactions (PPI enrichmentP< 0.001),indicating a particularly high degree of interaction between chaperone proteins (red cluster) and antioxidant proteins (green cluster).
Reactome and KEGG enrichment were applied to the data to gain further insight into reaction networks and relationships organized into biological pathways.The 153 significantly different proteins found in compa1 were analyzed.Seven pathways were enriched: five from KEGG (Table 2) and two from Reactome databases (Table 3).Importantly,the results appear to confirm an aberrant oxidative and ER response in the CMT1A rats compared to WT controls with emphasis on “Protein processing in the ER” and“Detoxification of ROS”.
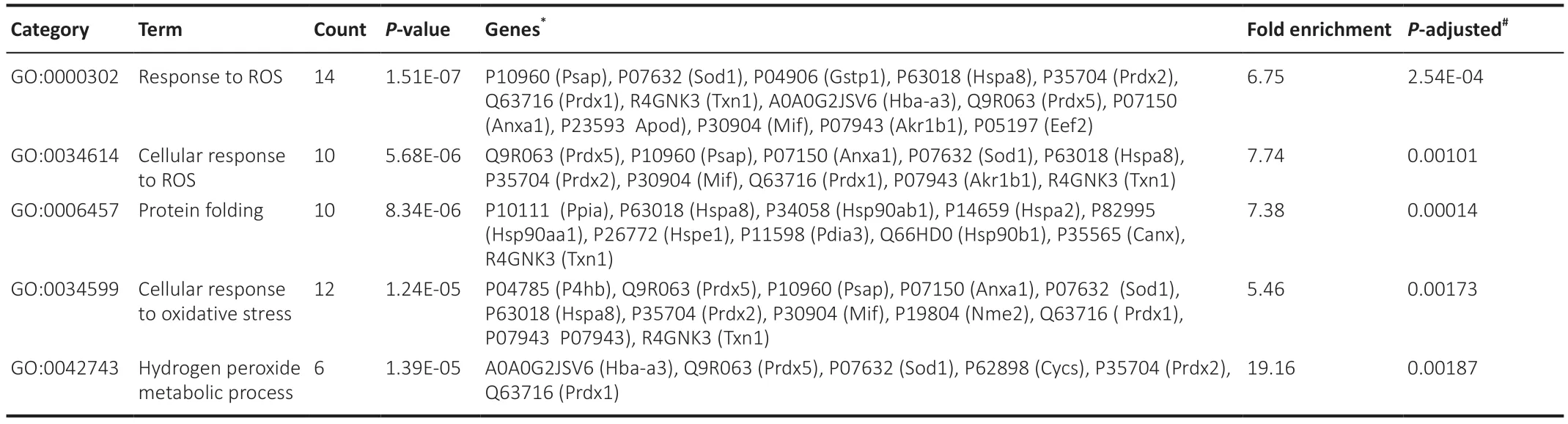
Table 1|Top five GO-BP terms filtered by fold enrichment > 5 (compa1) in order highest to lowest
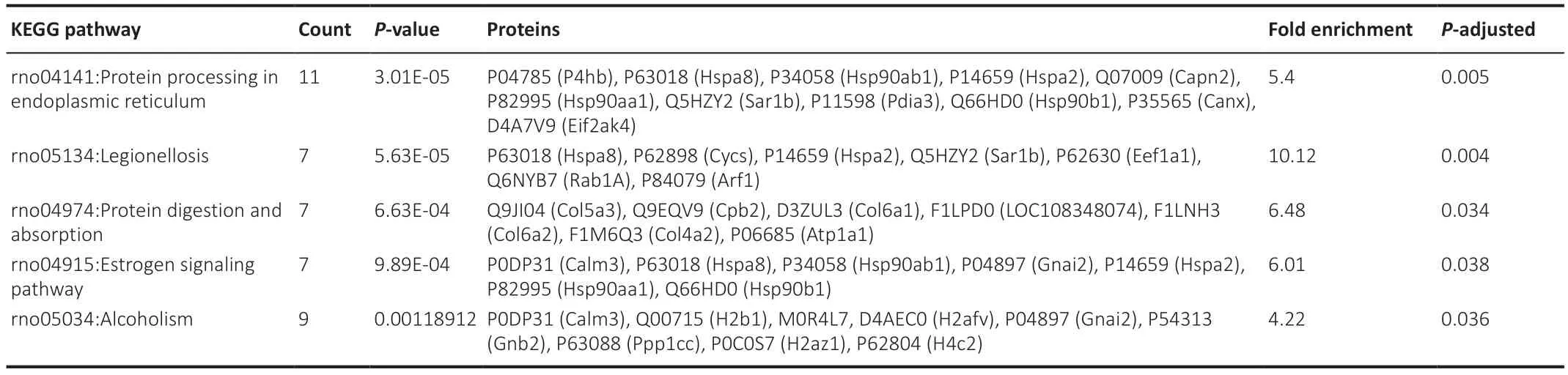
Table 2|KEGG pathways of compa1 (CMT1A versus WT)

Table 3|Reactome pathways of compa1 (CMT1A versus WT)
Target verification by western blotting: CMT1A versus WT
To confirm the validity of the proteomic results of compa1,the expression levels of 6 proteins were estimated by western blotting.These included 3 intracellular proteins (calnexin,peripherin,and Vimentin) that appeared to be over-expressed according to the proteomic results,and one potentially underexpressed (ALDR).In addition,PMP22 was evaluated.
Calnexin (CNX) is a type I transmembrane endoplasmic reticulum (ER) lectin that binds and folds proteins containing monoglucosylated oligosaccharides,such as PMP22 (Hammond et al.,1994).According to the proteomics data,CNX was overexpressed in each of the five CMT1A samples with an average FC=4.246.Due to its high level in CMT1A sciatic nerves,and its importance as an ER stress sensor,CNX was quantified by Western blotting (Figure 7A).CNX (90 kDa) was significantly overexpressed in CMT1A sciatic nerves (P<0.01;n=5) compared to WT (n=4).The blot inFigure 7Ais underexposed to highlight the upregulation of calnexin in the CMT1A nerves but on longer exposure,calnexin is easily detectable in WT nerves in line with other’s findings in the field (Fazal et al.,2017;Additional Figure 1).The average ratio of CNX quantification (CMT1A/WT)=5.472 (Figure 7B).
PMP22 protein,considered the major player in the pathology of CMT1A,was highly variable in CMT1A samples and less variable in WT samples,and no significant difference in PMP22 expression in the rat sciatic nerve was observed (Figure 7B).Very faint bands appeared at~100 kDa and~250 kDa(Additional Figure 2),which could represent PMP22 binding to chaperone proteins,but this point requires further confirmation.Peripherin (PERI,P21807) and vimentin (VIME,P31000),both type III intermediate filaments that assemble with other neurofilaments in neurons of the PNS,displayed a higher expression in the sciatic nerves of CMT1A rats with a ratio of CMT1A/WT of 8.717 for peripherin and 3.492 for vimentin (Figure 7AandB) in line with data from proteomic analysis.The oxidoreductase enzyme aldose reductase (ALDR,P07943) also known as aldo-keto reductase family 1 member B1,was involved in reducing carbonyl substrates such as sugar aldehydes and lipid peroxidation by-products (Reed,2011),was underexpressed in CMT1A in comparison to WT sciatic nerves in proteomic analysis(FC=0.486).Western blot analysis also showed that ALDR (37 kDa) had a lower expression (P< 0.05) in CMT1A samples (Figure 7AandB).
SNC versus WT (compa2)
In a parallel study,SNC was performed on WT rats.Injured rats showed signs of deterioration in sensorimotor function just after the crush injury,which improved with time (Caillaud et al.,2018).Sciatic nerves were collected and tested to explore their proteome 5 weeks after nerve injury.Out of the 459 proteins identified,92 proteins were significantly different (P< 0.05)compared to uninjured sciatic nerves from WT rats.As for compa1,PCA analysis was performed as a first visual exploration of the data and a 2D PCA plot of the 92 significantly different proteins was established (Figure 8A).As expected,the spread of data points is clearly separated for each group,indicating a difference in the protein expression profiles between groups,as seen in compa1 WT samples are more closely positioned,suggesting a similarity in the protein expression for the samples in this group.In contrast,SNC data points are more widespread,indicating greater heterogeneity in SNC samples.FC ratio of each protein (mean SNC/mean WT),indicated the majority of the altered proteins were downregulated in SNC samples compared to WT samples (Figure 8B) with 35 proteins downregulated and 2 upregulated in SNC groups (FC < 0.5 and > 2,respectively).These observations were also reflected in the heat map,showing that the majority of proteins were under-expressed in SNC groups compared to WT (Figure 9).Furthermore,protein interactions visualized by STRING (Figure 10) showed the PPI of the 92 affected proteins in compa2 was greater than expected for a random set of proteins (PPI enrichmentP< 0.001).
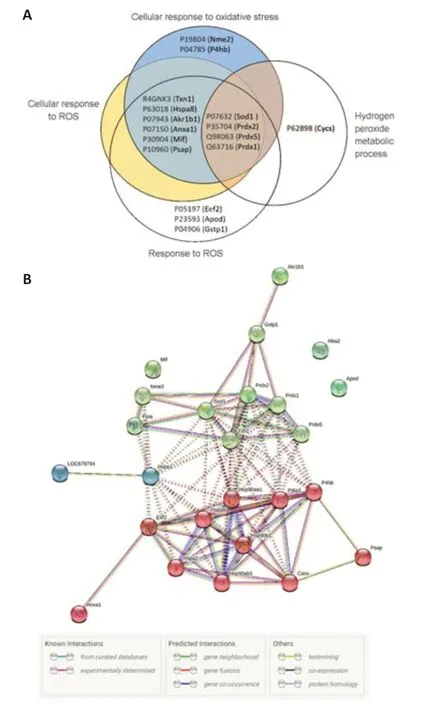
Figure 6|Analysis of the top five BP terms in compa1.
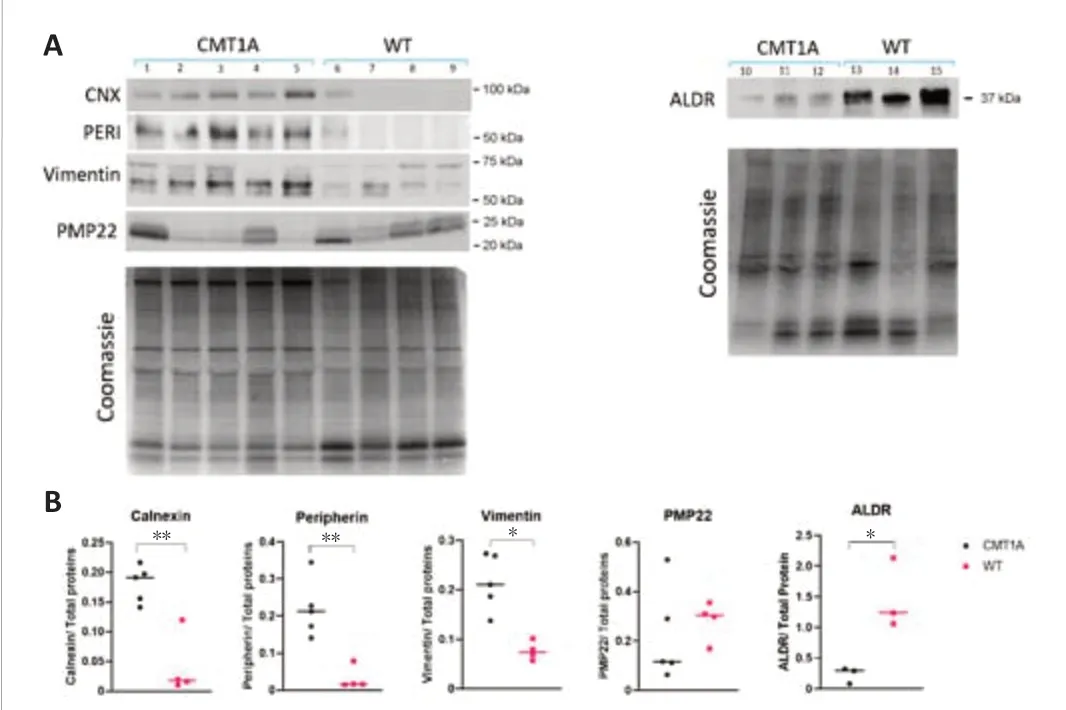
Figure 7|Protein expression analysis.
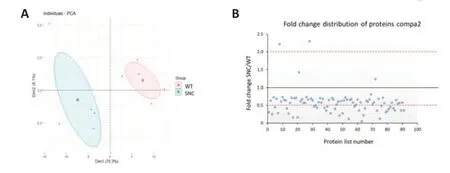
Figure 8|Data visualization of proteins sorted from compa2 (92 proteins).
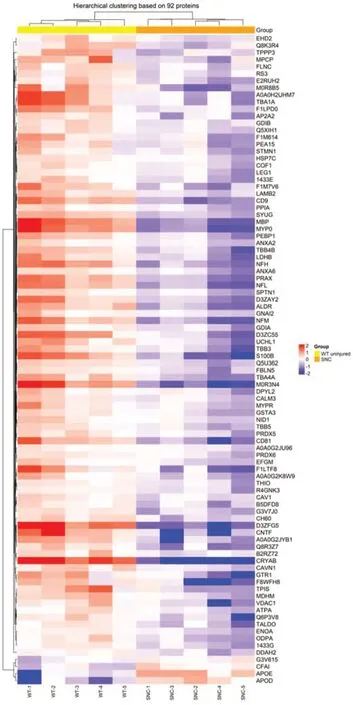
Figure 9|Heat map of SNC versus WT (compa2).
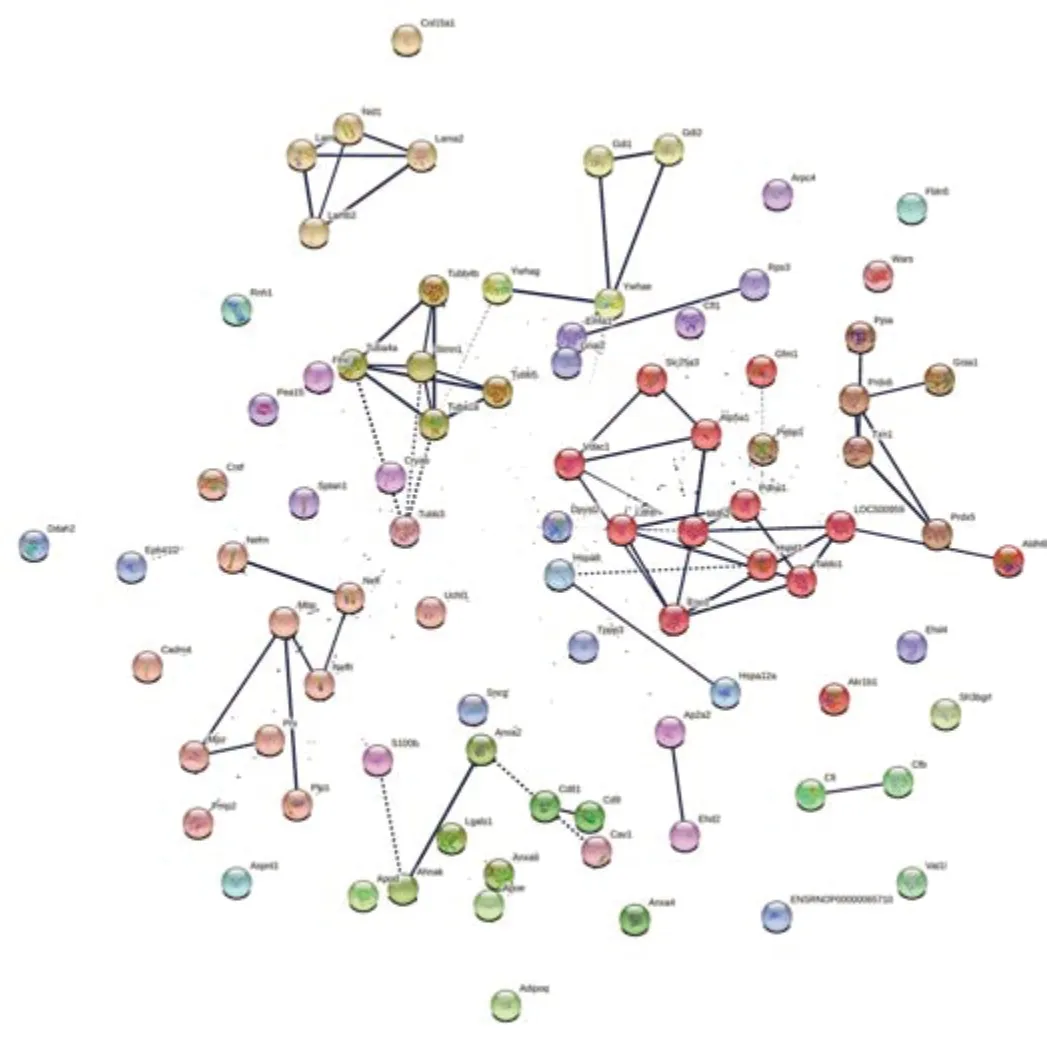
Figure 10|Protein network data of compa2 by STRING database (Rattus norvegicus).
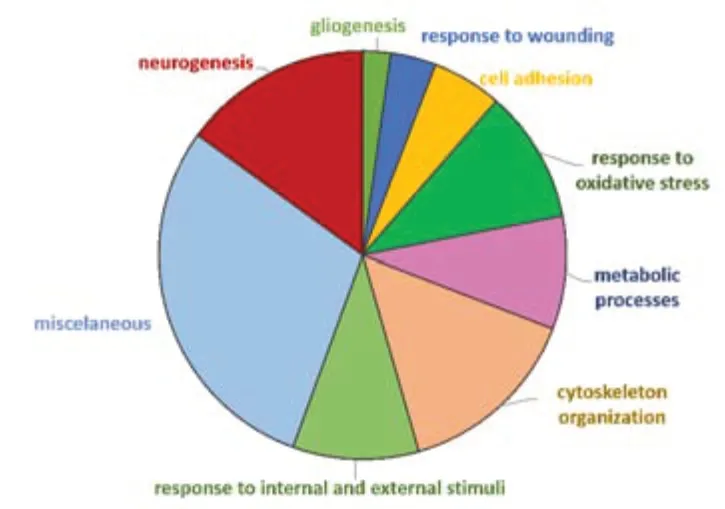
Figure 11|Pie chart of dysregulated proteins in SNC versus WT (compa2)annotated to Gene Ontology Biological Process (GO-BP) terms.
Similar to the approach followed for the previous analysis,upon further filtering of the GO data based onp-adjusted < 0.05 and fold enrichment >5,25 terms were highlighted (Additional Table 2).Then,the top-five terms were selected (Table 4).As expected,the most prominent terms in the SNC group were related to the response to axon injury (fold enrichment=18.49),including axon development and regeneration.Response to oxidative stress
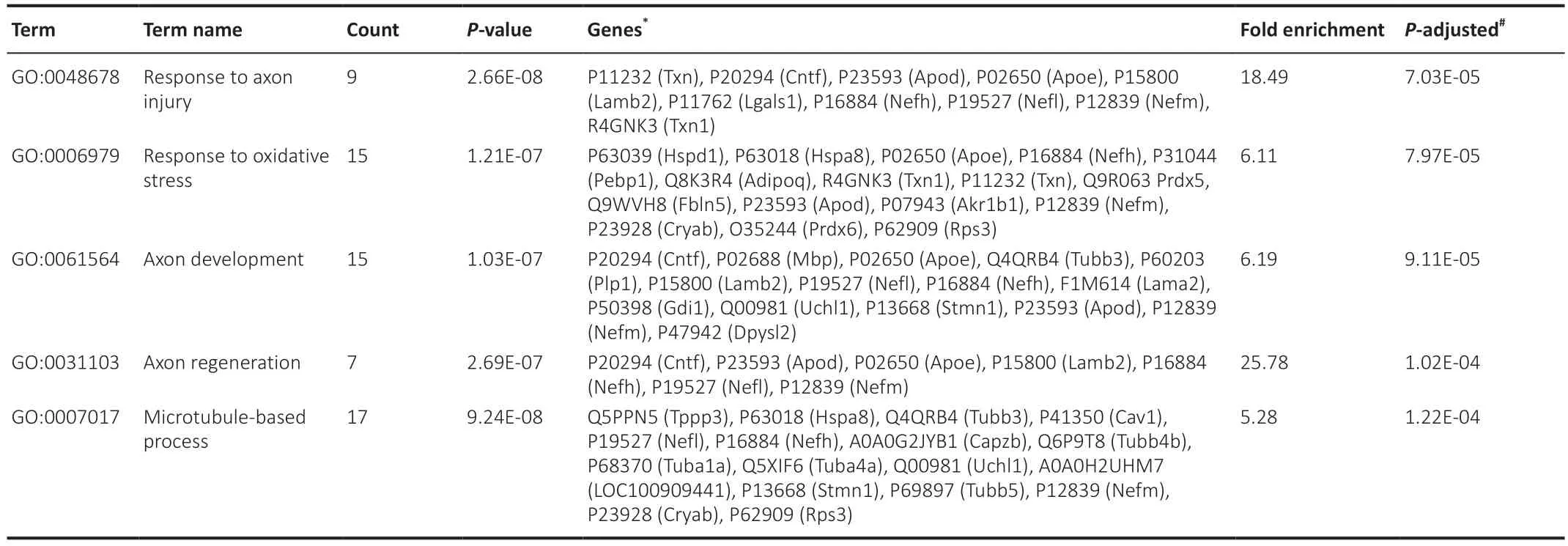
Table 4|Top five GO-BP terms filtered by fold enrichment > 5 (compa2) in order highest to lowest
was second (fold enrichment=6.11).Using the Venn-diagram method,18 irredundant proteins from the 3 terms related to axon injury/development were determined (Figure 12A).The common proteins between the 3 terms were: neurofilament heavy polypeptide (NFH,P16884),neurofilament medium polypeptide (NFM,P12839),neurofilament light polypeptide (NFL,P19527),apolipoprotein D (APOD,P23593),laminin subunit beta-2 (LAM-B,P15800),apolipoprotein E (APOE;P02650),and ciliary neurotrophic factor(CNTF,P20294).Oxidative stress-related proteins included thioredoxin,peroxiredoxins,ALDR,and alpha-crystallin B chain (P23928).The PPI of 17 proteins related to axonogenesis and 16 proteins related to oxidative stress were shown in Figure 12B.This network showed a significantly high level of interactions (PPI enrichmentP< 0.001).
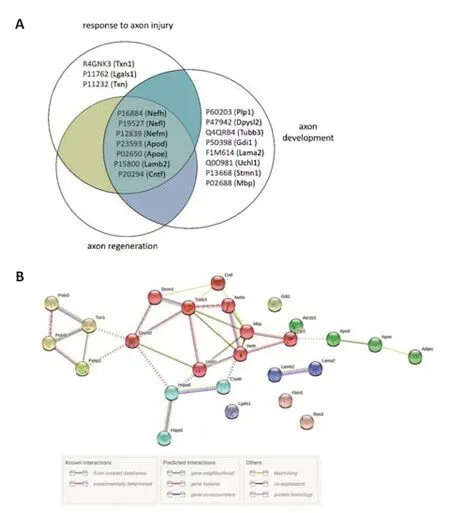
Figure 12|Analysis of the top five Biological Process terms in compa2.
Comparison of the two models (CMT1A versus SNC)
Finally,we investigated whether a protein signature might be shared between the two pathologies,CMT1A and SNC,which differ in origin,but result in similar symptoms.The differentially-expressed proteins in each of the two models were directly compared.A Venn diagram revealed that 32 proteins(13%) were dysregulated in common (Figure 13).Interestingly,these proteins varied in the opposite direction between the two comparisons (Table 5),i.e.proteins that were down-regulated in SNC samples (except APOD),were upregulated in CMT1A groups (except ALDR).Further filtering in compa1(CMT1Avs.WT) yielded 24 proteins with FC > 2 (Table 5),while in compa2,6 proteins had FC < 0.5 (Table 5,green).For example,NFL,which is involved in the maintenance of neuronal caliber,was increased three times in CMT1Avs.WT (compa1),while it decreased by two-thirds in SNC in comparison to WT (compa2).Moreover,PRDX5,which detoxifies peroxides and protects against oxidative stress,was increased almost two times in compa1,while it decreased to almost half in compa2 (Table 5).The two proteins APOD and ALDR that showed the opposite directions of change,had FC < 0.5 and >2,respectively (Table 5).APOD,a component of the high-density lipoprotein(HDL),has a very high fold increase in compa1 (FC=12.63) and a lower FC in compa2 (FC=2.304).ALDR,which plays a role in the transformation and detoxification of aldehydes and ketones,was substantially downregulated in both models when compared to WT (FC < 0.5).
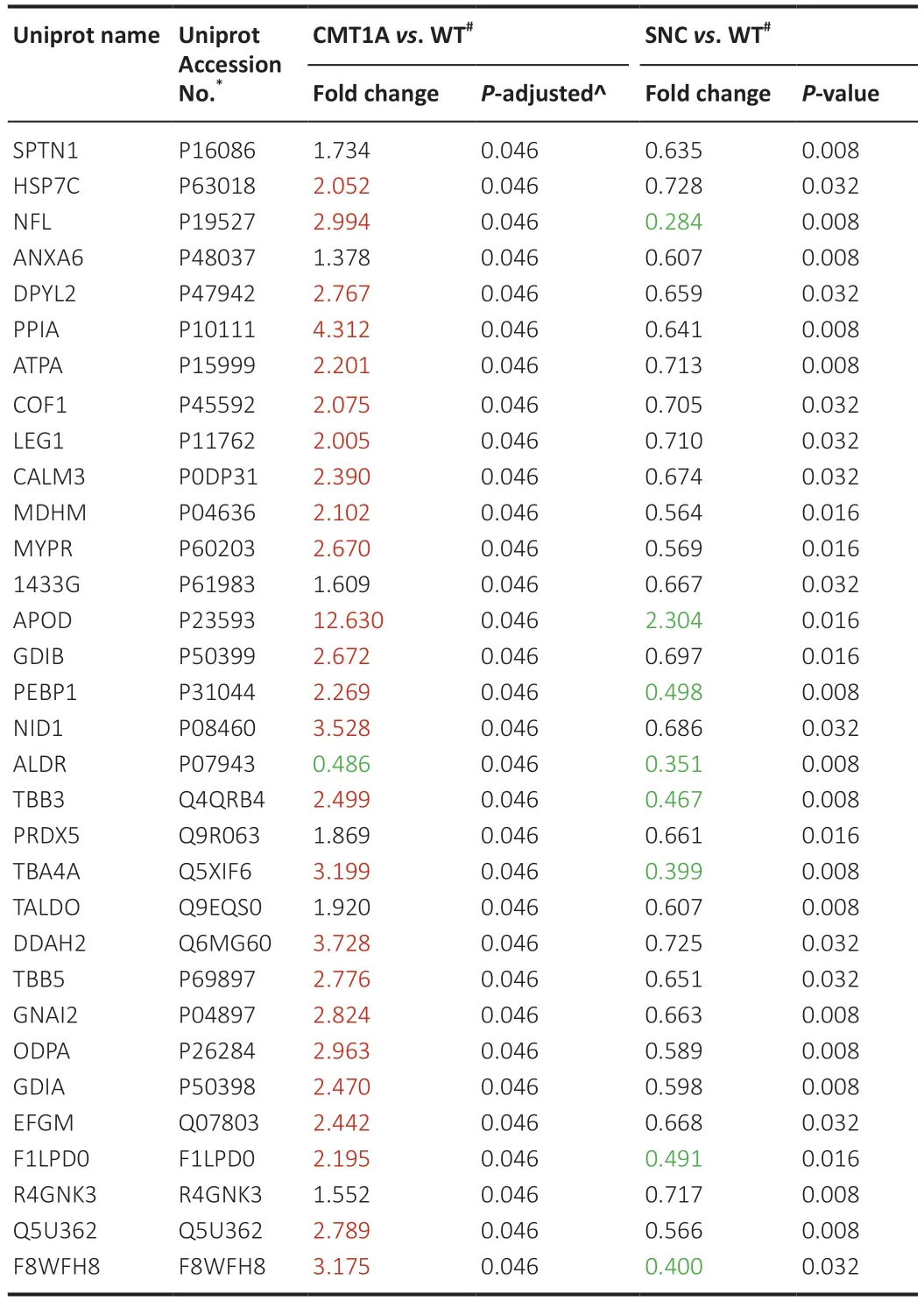
Table 5|Thirty-two deregulated proteins common to both compa1 and compa2
The two GO-BP terms lists generated from compa1 and compa2 were compared.Forty-four BP terms were found to be in common,54 terms were specific to compa1 and 28 terms were specific to compa2 (Figure14A).Among the 44 common terms,17 terms were filtered according toP-adjusted < 0.01 in the two enrichment analyses (Figure 14B).As expected,the prevalent terms were related to neurogenesis and oxidative stress.To further parse the data,only proteins that had a > 4-fold enrichment in the two enrichment analyses were selected.Out of the 44 terms,only 12 terms met the criteria,all of which were relevant to metabolic processes as well as response to oxidative species and axon injury (Figure 14C).
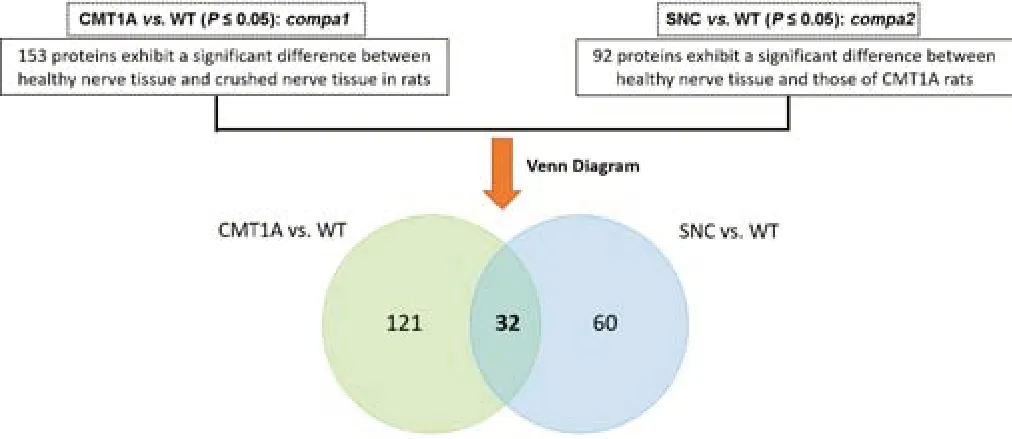
Figure 13|Venn diagram of dysregulated proteins common to both compa1 and compa2.
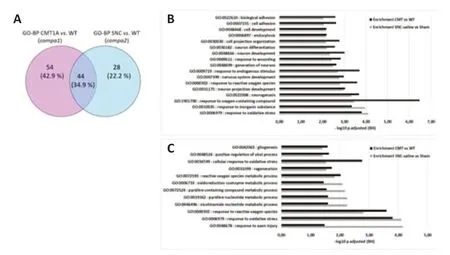
Figure 14|Overlap of GO-BP lists between compa1 and compa2.
Discussion
SWATH-MS technique was used to determine the protein signature in CMT1A and SNC rat models,extending our previous studies (Caillaud et al.,2018,2020).We aimed to examine how cells of the sciatic nerve respond to the disease processes of genetic and traumatic peripheral neuropathy.Indeed,the change in protein expression strictly correlates with the age and stage of the disease,implying a possible difference in protein signature if a differenttime point was to be chosen.At three months old,sensorimotor symptoms of CMT1A rats are well-established.Moreover,5 weeks after crush injury,rats showed partial functional and histological repair.A comparison of the two conditions would reveal potential common variations linked to PN and provide insight into potential targets for future therapeutic approaches.
When coming to each study,high-dimensional data,where the number of features exceeds the number of observations,is common in biology and arises when multiple features such as the expression of many genes,are measured for each sample (Lever et al.,2017).Principal component analysis(PCA) is an efficient method to reduce a large number of variables to a smaller number of groups that can be more readily visualized and understood(Ivosev et al.,2008).It allows patterns to be found without reference to prior knowledge about whether the samples come from different treatment groups or have phenotypic differences (Lever et al.,2017).To interpret the functional significance and biological output of the altered proteins,enrichment for different biological terms (according to Gene Ontology terms,KEGG,and Reactome pathways) was used.These bioinformatics tools help to visualize,interpret and analyze data with respect to biological pathways and relevance to basic and clinical research.
In the first comparison (CMT1AversusWT),the majority of identified proteins were significantly upregulated in CMT1A samples.Several GO terms emerged consistent with CMT1A physiopathology,such as detoxification,response to oxidative stress,neurogenesis,and protein folding.Response to oxidative stress was a pivotal emerging term.It appears to be a chief feature of CMT1A disease,potentially playing a role in the pathology.We have recently demonstrated the presence of oxidative stress in CMT1A rats,manifested by high ROS production in sciatic nerves and lipoperoxidation markers in urine (Caillaud et al.,2020).Sorted proteins also included SOD and GST,both of which have been documented to be elevated in the plasma of CMT1A patients (Chahbouni et al.,2017).
Consistent with the toxic gain of function of the PMP22 gene in CMT1A disease,“protein folding” emerged as an important term.Sorted proteins were generally molecular chaperones.In particular,five HSPs were identified:HSC70,HSP90α,HSP90β,GRP94,HSP70-2,and HSP10.These chaperones are responsible for correct protein folding during physiological conditions and for restoration,refolding,and preventing aggregation of damaged or denatured polypeptides in cells exposed to stress (Gupta et al.,2020).In the context of CMT1A,overexpression of these chaperones was expected.However,whether the protein overload aggravates the injury over time needs further study.CNX is an ER chaperone that binds and folds glycoproteins such as PMP22.It is also a direct protein folding sensor (Hammond et al.,1994).Western blotting demonstrated that CNX was highly expressed in CMT1A samples,compared with WT samples,concordant with proteomic data.One mechanism of ER quality control checkpoints is the CNX/calreticulin cycle that promotes protein folding.In our study,calreticulin (CALR,P18418),the luminal counterpart of CNX,was identified by MS but did not vary significantly from WT.In accordance with our study,Jung and colleagues showed that CNX,but not CALR,plays a crucial role in the folding and function of PMP22(Jung et al.,2011).Thus,CALR would not appear to be a major player in the pathophysiology of CMT1A.High expression of CNX in CMT1A rats may be a compensatory mechanism to facilitate the folding/refolding of PMP22 and alleviate the burden of misfolded proteins within the ER (Caillaud et al.,2020).However,this mechanism would seem to be insufficient to prevent demyelination and the consequent symptoms of CMT1A.
Terminally-misfolded glycoproteins are targeted for degradation by the ERAD pathway.In contrast,an unfolded protein response (UPR) is activated when misfolded proteins accumulate (Hwang and Qi,2018).If the condition is not resolved,UPR may lead to cell death.The ER chaperone BiP is a primary activator of UPR (Kopp et al.,2019).While most ERAD factors identified in our study (CNX,GRP94,proteasome subunit-alpha,and UBC) were overexpressed in CMT1A,BiP was not changed.We hypothesize that it is rather ERAD and not UPR that is activated at this time-point of the disease.On the other hand,PMP22 was tested by western blot because it was not identified by MS.Its expression was variable,most notably in CMT1A samples.In fact,PMP22 has a rapid turnover rate,with a half-life of approximately 70 minutes (Ryan et al.,2000).Therefore,a large portion of newly synthesized PMP22 is degraded,and only a small portion is transported to the cell surface (Li et al.,2013).Our observations may be explained by inter-individual variability,being further perturbed by CMT1A disease.
At the axonal end,high expression of neurofilaments,such as NFL and peripherin,as well as the intermediate filament vimentin,indicates modulation of axon growth.Since it is known that the interaction between Schwann cells and the axon is essential in both physiological and pathological cases,hypomyelination observed in CMT1A models and patients (Jouaud et al.,2019) is expected to affect the axon at a certain point of the disease (Maier et al.,2002).In CMT1A rats aged one year,decreased axon caliber was already observed (Gautier et al.,2021).However,in CMT1A rats aged 3 months,we hypothesize that Schwann cell perturbations are accompanied by mechanisms to preserve the axon,manifested by increased expression of neurofilaments.Interestingly,several neurofilament proteins are used as blood biomarkers to monitor neurological diseases and the efficacy of therapies (Yuan and Nixon,2021).ALDR is involved in the detoxification of reactive aldehydes such as the cytotoxic by-product of lipid peroxidation 4-hydroxynonenal.It has been shown that these reactive aldehydes are increased in several neurodegenerative diseases (Reed,2011).Although aldehydes are generated in neurons and Schwann cells via normal metabolic processes,the production of reactive aldehydes is increased in cells exposed to high levels of oxidants.As oxidative stress is already proven to occur in CMT1A sciatic nerve (Caillaud et al.,2020),high levels of reactive aldehydes could be a contributing source.Therefore,ALDR may represent an important detoxification route within SCs.In our case,the low level of ALDR in CMT1A sciatic nerves may lead to the accumulation of reactive aldehydes and thus contribute to oxidative stress.
On the other hand,SNC is a traumatic nerve damage model with a relatively high clinical incidence.In compa2 where SNC was compared to WT,most proteins at 5 weeks post-injury were significantly under-expressed in SNC in comparison to WT sciatic nerves.Identified GO terms included neurogenesis,gliogenesis,cytoskeletal organization,as well as response to wounding and oxidative stress.Peripheral nerve injury induces cellular and molecular changes at the level of neurons,Schwann cells,as well as in the local microenvironment.These changes are first accompanied by anterograde Wallerian degeneration and the formation and proliferation of“repair” Schwann cells.This is followed by a regenerative phase,where the microenvironment becomes permissive for axon regrowth and remyelination(Jessen and Mirsky,2019;Arthur-Farraj and Coleman,2021).Despite the known proliferation of Schwann cells after injury,myelin proteins MBP and PLP were under-expressed.Moreover,alpha-crystallin B chain and periaxin,highly expressed in myelinating Schwann cells,were strongly downregulated.In fact,alpha-crystallin B chain and periaxin have been shown to be downregulated soon after SNC and downregulation persists for as long as 35 days (Jiménez et al.,2005).This decrease in myelin proteins should be seen in the scope of the phenotypic changes in Schwann cells after injury.Neural cytoskeletal proteins NFH,NFM,and NFL,as well as other cytoskeleton-related proteins such as laminin,tubulin,cofilin-1,stathmin,and spectrin were downregulated.The change in axon proteins could be linked to the decrease in axon caliber at this time point after injury.This concurs with our previous nerve conduction velocity and myelination tests on the same animals which show a persistent injury profile,even though sensorimotor functions were improved (Caillaud et al.,2018).Prolonged low levels of myelin and axon proteins may indicate a defect in the repair machinery.
In addition,we identified several proteins that regulate regeneration including ciliary neurotrophic factor (CNTF) and caveolin-1 (CAV1),both of which were decreased in SNC.CNTF is implicated in preventing the degeneration of motor axons after axotomy (Park et al.,2004).In contrast,CAV1 knockout mice revealed remarkably high levels of vascular endothelial growth factor and the formation of new neurons (Li et al.,2011).Thus,the interplay between proand anti-regenerative factors determines the post-injury outcomes.During nerve regeneration phase,lipids are essential for remyelination.Consistent with this,APOD and APOE were increased after SNC.Expression of antioxidant enzymes such as PRDX 5,6 and THIO was decreased expression.HSPs such as HSP70 and mitochondrial HSP60,appeared to be shut down at this time point(5 weeks) post-injury.Inflammatory proteins annexin A2,annexin A6,and S100B were downregulated.These proteins have been previously reported to form complexes that have a wide array of functions including inflammation(Weisz and Uversky,2020).Furthermore,annexin A2 is involved in promoting the proangiogenic phenotype in macrophages (Wang et al.,2018).
Taken together,the results of the present study are complementary to other studies on CMT1A and SNC models and give insight into protein perturbation in peripheral nerve injury.CMT1A is a slowly progressive disease that tends to worsen over time.In contrast,depending on the severity of the physical injury,SNC models tend to improve with time.Our data highlight the presence of shared disturbances in cellular processes,regardless of the direction of change,i.e.either up-or down-regulation.Common processes in both models include oxidative stress and response to axon injury.A bigger sample size of animals could give a more precise idea of the protein variation and of in case of heterogeneity in the population.Furthermore,to gain specific insights into the myelin proteome,myelin purification could be useful as has been recently performed in mice (Siems et al.,2020).Time-dependent details of protein expression changes,as recently published by Aiki and colleagues for SNC(Aiki et al.,2018),would be desirable.Nevertheless,the present findings,at these time points,provide a group of divergent proteins that could be further investigated either to ameliorate the characterization of peripheral nerve repair/regeneration or to search for novel therapeutic targets.
Acknowledgments:The authors are indebted to Prof.Klaus-Armin Nave and Prof.Michael Sereda for the CMT1A rats supplied by the Max Planck Institute for Experimental Medicine (University of Göttingen,Göttingen,Germany).
We thank the staff of the animal house facility at the Faculty of Medicine and Pharmacy,Limoges.We thank Dr.Hassan Aouad (INSERM,UMR 1248,University of Limoges,Limoges,France) for his advice during the data analysis process.We thank Dr.Ian Darby (Editingbiomed,Melbourn,Australia) for the professional editing of the manuscript.
Author contributions:ZM analyzed the data,performed western blot experiments and STRING analysis,and wrote the original manuscript draft.SD performed statistical and bioinformatics analysis.EP performed the proteomics experiment.MC performed the sciatic nerve crush experiment.
MC and LV collected the sciatic nerves.AD,MEM,and FB provided supervision throughout the work.AD,MEM,and SD reviewed the manuscript.All authors approved the final submitted version.
Conflicts of interest:The authors declare no conflicts of interest.
Availability of data and materials:All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:
Additional Table 1:Gene Ontology-Biological Process terms of CMT1A versus WT (compa1) filtered by statistical significance and fold enrichment > 5.
Additional Table 2:Gene Ontology-Biological Process terms of SNC versus WT(compa2) filtered by statistical significance and fold enrichment > 5.
Additional Figure 1:Overexposed calnexin western blotting.
Additional Figure 2:PMP22 western blotting.

- 中国神经再生研究(英文版)的其它文章
- Neuro faces of beneficial T cells: essential in brain,impaired in aging and neurological diseases,and activated functionally by neurotransmitters and neuropeptides
- Profiling neuroprotective potential of trehalose in animal models of neurodegenerative diseases:a systematic review
- Cdk5 and aberrant cell cycle activation at the core of neurodegeneration
- Recent advancements in noninvasive brain modulation for individuals with autism spectrum disorder
- Vicious cycle of lipid peroxidation and iron accumulation in neurodegeneration
- Cell-based therapeutic strategies for treatment of spinocerebellar ataxias: an update