
Cdk5 and aberrant cell cycle activation at the core of neurodegeneration
2023-01-21 04:42RaquelRequejoAguilar
中国神经再生研究(英文版) 2023年6期
Raquel Requejo-Aguilar
Abstract Neurodegenerative diseases are caused by the progressive loss of specific neurons.The exact mechanisms of action of these diseases are unknown,and many studies have focused on pathways related to abnormal accumulation and processing of proteins,mitochondrial dysfunction,and oxidative stress leading to apoptotic death.However,a growing body of evidence indicates that aberrant cell cycle re-entry plays a major role in the pathogenesis of neurodegeneration.The activation of the cell cycle in mature neurons could be promoted by several signaling mechanisms,including c-Jun N-terminal kinases,p38 mitogen-activated protein kinases,and mitogen-activated protein kinase/extracellular signal-regulated kinase cascades;post-translational modifications such as Tau-phosphorylation;and DNA damage response.In all these events,implicated Cdk5,a prolinedirected serine/threonine protein kinase,seems to be responsible for several cellular processes in neurons including axon growth,neurotransmission,synaptic plasticity,neuronal migration,and maintenance of neuronal survival.However,under pathological conditions,Cdk5 dysregulation may lead to cell cycle re-entry in post-mitotic neurons.Thus,Cdk5 hyperactivation,by its physiologic activator p25,hyper-phosphorylates downstream substrates related to neurodegenerative diseases.This review summarizes factors such as oxidative stress,DNA damage response,signaling pathway disturbance,and Ubiquitin proteasome malfunction contributing to cell cycle re-entry in postmitotic neurons.It also describes how all these factors are linked to a greater or lesser extent with Cdk5.Thus,it offers a global vision of the function of cell cycle-related proteins in mature neurons with a focus on Cdk5 and how this protein contributes to the development of Alzheimer’s disease,Parkinson’s disease,amyotrophic lateral sclerosis,and Huntington’s disease by cell cycle activation.
Key Words: Alzheimer´s disease;amyotrophic lateral sclerosis;apoptosis;Cdk5;cell cycle;Huntington´s disease;neurodegeneration;neuron;oxidative stress;Parkinson´s disease;signaling;Tau phosphorylation
From the Contents
Introduction 1186
Search Strategy and Eligibility Criteria 1186
Factors Contributing to Neuronal Cell Cycle Re-entry 1186
Role of Cell Cycle Proteins in Neurons 1187
Cdk5 and Cell Cycle Activation in Neurons 1188
Cdk5 Dysfunction in Neurodegenerative Disorders 1188
Conclusions 1189
Introduction
Neurons,because of their specialized functions,remain quiescent once differentiated bearing with it a strict regulation of the cell cycle.This regulation lies with a group of proteins,cyclins,and cyclin-dependent kinases (CDKs) that control specific cell cycle checkpoints which determine its progression (Joseph et al.,2020).Cell cycle regulatory proteins are also important in mature neurons since they have other functions including axon guidance,synaptic function,and plasticity control (Akagawa et al.,2021).However,some of these proteins might get dysregulated and activated in response to pathological conditions which lead to cell cycle re-entry (Gupta et al.,2021).Furthermore,neurodegenerative diseases are characterized by the aggregation of non-functional proteins such as amyloid-β,p-Tau,α-synuclein,huntingtin,and parkin (Sampaio-Marques et al.,2019;Chao et al.,2020;Park and Barrett,2020).These aggregates cause physiological changes in cells that trigger several signaling pathways including mitogen-activated protein kinase (MAPK),c-Jun N-terminal Kinases (JNK),Ras/Raf/mitogen-activated protein kinase/ERK kinase/extracellular-signal-regulated kinase (MEK/ERK)and apoptotic pathways.Taken together,increased expression of cell cycle markers and pathological protein deposition strengthen the case that cell cycle activation is the very starting point for neurodegeneration (Farmer et al.,2020).The final consequence of cell cycle re-entry in mature neurons is apoptotic cell death,a common feature in neurodegenerative disorders.In addition,there are other processes common to them and several studies have demonstrated the implication of oxidative stress,mutations,mitochondrial dysfunction,and impairment of proteasome pathway and protein aggregates in cell cycle re-entry in mature neurons (Tripathi et al.,2021).In response to a high concentration of reactive oxygen species (ROS) and DNA damage,mitogenic signals are activated,inducing DNA replication and cell cycle,to promote DNA repair.However,in post-mitotic neurons,DNA damage response (DDR) is defective and is associated with neurodegeneration (Lou et al.,2021).DNA damage also affects mitochondria,impairing ATP production,which activates AMP-activated protein kinase (AMPK).AMPK stimulation is involved in defects at G1/S transition and G2/M arrest (Joseph et al.,2020).
Of all cell cycle kinases,cyclin-dependent kinase-5 (Cdk5) is an atypical kinase with a minimal role in cell cycle regulation under normal conditions.However,Cdk5 is especially important in suppressing cell cycle re-entry in post-mitotic neurons which occurs in several neurodegenerative diseases (Gupta et al.,2021;Lopez-Grueso et al.,2022;Pandey and Vinod,2022).Cdk5 can act directly by suppressing the cell cycle in neurons through disruption of the E2F1-DP1 complex.Cdk5 pulls up E2F1 from DP1 and forms a new complex of three proteins along with p35.p35 and p39 are regulatory subunits of Cdk5 that are essential for the correct function of this kinase including cell cycle suppression.Aberrant Cdk5 activation and induction of the cell cycle happen when a truncated version of p35 (p25) is produced.Cell cycle reentry through complex Cdk5-p25 also occurs after the interaction between p25 and histone deacetylase 1 (HDAC1).HDAC1 is a transcriptional repressor of cell cycle genes and when attached to p25 and inactivated,it increases cell cycle gene expression and double-stranded DNA breaks leading to neuronal death (Allnutt et al.,2020).Cdk5 may also act indirectly over the cell cycle,for instance,Cdk5 modulates p53 levels and activity.Thus,Cdk5/p25 complex increases p53 levels,and its phosphorylation at Ser15 contributes to cell cycle arrest in the G2/M phase and increases in apoptosis in response to damaging stimuli such as amyloid-β accumulation and exposure to the carcinogenic pollutant benzo[a]pyrene (Lapresa et al.,2019;Nie et al.,2022).
Search Strategy and Eligibility Criteria
Literature search and selection processes were carried out between April and July 2022.The databases used were PubMed,Research Gate,and Google Scholar,and only literature from the last 5 years with few exceptions was selected.Searched terms were “Mature or postmitotic neurons AND Cell cycle or cell cycle activation”,“Cdk5 AND Neurons”,“Aberrant cell cycle re-entry factors”,“Proteasome AND Cell cycle activation”.The results were further screened by reading the abstract.
Factors Contributing to Neuronal Cell Cycle Re-entry
There are several processes such as aging along with environmental and genetic factors that contribute to the development of neurodegenerative disorders by affecting proteasomal system function,altering molecular pathways,DNA and mitochondrial damage and ROS increase (Figure 1).
Oxidative stress
The mechanism of cell death in neurodegenerative diseases caused by cell cycle re-entry in neurons is related to an increase in ROS and oxidative stress that could be both cause and consequence(Figure 1).Alzheimer´s disease(AD),Parkinson´s disease (PD),amyotrophic lateral sclerosis (ALS),and Huntington´s disease (HD) show molecular markers of both cell cycle re-entry and oxidative stress (Oli et al.,2021).Oxidative stress can cause DNA damage and malfunction of DNA repair mechanisms,which leads to mitochondrial dysfunction,another hallmark of neurodegenerative diseases which in turn increases ROS generation,creating a vicious cycle.Mitochondrial dysfunction decreases ATP production and produces a metabolic shift from oxidative phosphorylation (OXPHOS) to glycolysis that has been observed in PD and other neurodegenerative diseases (Atlante et al.,2017;Vallee et al.,2018;Bell et al.,2020).For instance,Hexokinase 2 and other glycolytic enzymes are upregulated in the MPTP-induced mouse model of PD and SH-SY5Y cells treated with MPP+,increasing lactate levels and apoptotic cell death by activation of the AMPK/Akt/mTOR pathway (Li et al.,2022).Furthermore,activation of AMP-activated protein kinase (AMPK) prevents the transition of cells from G1 to S phase,affecting the G1/S checkpoint;when mitochondrial dynamics are altered,regulatory proteins involved in G2/M arrests,such as cyclin A,cyclin B1,CDK1,polo-like kinase1 (Plk1),aurora kinase A and Mad2,are increased,leading to neuronal apoptosis (Joseph et al.,2020).Moreover,the increases of ROS and mitochondrial dysfunction upregulate cyclin D and retinoblastoma protein (pRb) levels,activating the cell cycle(Joseph et al.,2020).Thus,neurotoxins such as 3-nitropropionic acid (3-NP)that inhibits succinate dehydrogenase and impairs mitochondrial function enhance cyclin D1 expression and cell cycle re-entry that cause neuronal death in the striatum (Dietrich et al.,2022).Higher levels of cyclin D1 have also been observed in cortical neurons exposed to topoisomerase I inhibitor camptothecin which induces DNA damage and oxidative stress,leading to cell cycle-related death (Zhang et al.,2020b).Furthermore,increased cyclin D1 and evidence of cell cycle re-entry are observed in most neurodegenerative diseases (Dietrich et al.,2022).Oxidative stress and aberrant cell cycle are also linked by activation of mitotic signaling cascades,DNA damage response,and impairment of unfolded protein response (Figure 1),which will be discussed below.
DNA damage response
ROS induce DNA damage in a variety of ways,including the formation of double-strand breaks (DSBs),the most lethal forms of DNA damage,that are associated with neuronal loss on a broad range of neurodegenerative diseases(Mitra et al.,2019;Thadathil et al.,2021;Hinkle et al.,2022;Konopka and Atkin,2022).To prevent the effects of oxidative stress-induced DNA damage,cells have developed a damage response mechanism (DDR) that requires cell cycle re-entry to be initiated (Figure 1).Thus,activation of DDR by oxidative DNA damage in post-mitotic neurons is a protection and repair mechanism.However,a persistent oxidative DNA damage becomes detrimental since it involves cell cycle activation and neuronal death (Provasek et al.,2022).In response to DSBs,essential elements of G0/G1 transition,such as cyclin D1,pRb and DNA helicase subunit mini-chromosome maintenance protein 2(Mcm2),are activated in differentiated neurons.Conversely,DDR decreases when components of G1 transition such as CDK4 and CDK6 are suppressed(Joseph et al.,2020).Neuronal death,through cell cycle activation and DDR response,depends on cell cycle checkpoints and ataxia telangiectasia mutated (ATM) is a key kinase of the response to DSBs that transduces damage signals to these checkpoints.ATM suppresses cell cycle re-entry in post-mitotic neurons and its decreases are linked to neurodegeneration.ATM levels are reduced in human frontal cortex and cerebellum from AD patients and its activity in neurons is low as indicated by the translocation of histone deacetylase 4 into the nucleus,trimethylation of histone H3 and increases of cell cycle proteins such as cyclin A2 (Pizzamiglio et al.,2020).
Dysregulation of ATM has been also described in cells transfected with a mutated form of Huntingtin that showed increased ATM activation.The same was observed in mouse models and brain tissue from patients with HD and mouse models of HD (Pizzamiglio et al.,2020).In ALS cells and animal models,activation of ATM,and through it,checkpoint kinase 2 (CHK2) and p53 cause a chronic activation of DDR and increase neuronal death (Yang et al.,2020b).Despite all these data,the interaction of DNA damage with DDR and neuronal cell cycle activity at molecular levels are still not fully elucidated.
Molecular pathways implicated in neuronal cell cycle re-entry
Evidence suggests a connection between the cell cycle and different biological processes such as apoptosis,autophagy,inflammation,and mitochondrial function with the involvement of several signaling pathways (Figure 1).For instance,PI3K/AKT/GSK3b is one of the most important pathways for neuronal survival and is involved in neuroinflammation.Activation of Akt requires its phosphorylation at the Ser473 site and GSK3b is inactivated when it is phosphorylated at Ser9 by Akt activation.When activated,GSK3b increases inflammation by increasing proinflammatory cytokines via NFkB and Tau phosphorylation.Further,an increase in active GSK3b has been described in brain samples of patients with AD (Yang et al.,2020a).Sulforaphane treatment upregulates the p-Akt (Ser473)/Akt ratio and inhibits the activation of GSK3b,which improve cognitive function and reduce AD in animal models of this pathology (Yang et al.,2020a).Moreover,activation of GSK3b/b-catenin promotes neurogenesis and chronic fluoride administration inhibits this pathway and reduces the expression of β-catenin downstream targets c-Myc and cyclin D1,suppressing cell cycle progression (Jiang et al.,2019).Thus,GSK3b inhibition might be a good strategy for the treatment of neurodegenerative diseases.

Figure 1|Factors related to cell cycle re-entry in neurons.
MAPKs,including ERK/MEK,p38 MAPKs,and JNKs,are signaling molecules that regulate cellular activities,including cell survival/death and proliferation/differentiation.The binding of mitogens,like epidermal growth factor and brain-derived neurotrophic factor,to the cell surface,leads to ERK phosphorylation and translocation to the nucleus which regulate the transcription of genes involved in cell proliferation.Further,JNK and p38 MAPK,also known as stress-responsive MAPKs,are involved in cell proliferation under pathological conditions.The overwhelming evidence is that abnormal phosphorylation and activation of MAPKs are related to neuronal death and neurodegeneration.For instance,the p38 MAPK cascade is activated by both Aβ peptide and the presence of tauopathies contributing to memory deficits in mouse models of AD (Falcicchia et al.,2020).Activation of microglial MAPKs has also been related to neuronal death in PD,HD,and ALS models (Gil-Martinez et al.,2020;Li et al.,2020;D’Mello,2021;Yadav et al.,2021).Thus,inhibition of these proteins has been reported to attenuate COX-2,iNOS,microglial hyperactivation,and ROS in these pathologies and improves cell viability (Kwon and Lee,2020;Li et al.,2020;D’Mello,2021;Sahu et al.,2021).
Moreover,activation of ERK,p38,and JNK pathways induces apoptosis of PC12 cells through mitochondrial dysfunction.Activation of the MAPK pathway decreases Bcl2 expression and promotes Bax translocation to the mitochondria which in turn activates caspase-3,leading to mitochondrial dysfunction and apoptotic cell death (Zhang et al.,2020a).
Ubiquitin proteasome system
The cell cycle is regulated directly by cyclin-dependent kinases (CDKs),CDK inhibitors (CKIs),and protein degradation by the ubiquitin-proteasome system (UPS).The levels of CDK-cyclin complexes and CKIs oscillate during the cell cycle because of the action of the ubiquitin ligases (SCF complexes and APC/C) and proteolysis (Roy et al.,2022).Abnormalities in UPS function associated with neurodegenerative disorders not only promote accumulation and aggregation of damaged or misfolded proteins such as amyloid-β,tau,α-synuclein,fused-in-sarcoma,or HTT but also promote the activation and progression of the abnormal cell cycle.Thus,UPS dysfunction affects the concentration of cyclins,CDKs,CKIs,tumor-suppressor proteins,such as p53,and transcription factors altering cell cycle control (Roy et al.,2022).Thus,the anaphase-promoting complex or cyclosome (APC/C) is involved along with its activating proteins Cdc20 and Cdh1 in the exit from mitosis (APC/CCdc20)and maintenance of the G1 phase (APC/CCdh1).However,under pathological conditions,both forms of this ligase are involved in the activation of cell cycle checkpoints.Further,in vitroandin vivomodels of AD show an increase in Cdh1 phosphorylation,leading to the inactivation of APC/C and neuronal apoptosis (Lapresa et al.,2022).SCF (Skp1-Cullin-F-box protein) complexes are formed by S-phase kinase-associated protein 1 (Skp1),ring-box 1 (Rbx1),Cullin 1 (Cul1),and a variable F-box protein.F-box and WD repeat domain containing 7 (FBXW7) is one of the components of SCF E3 ubiquitin ligases and has been involved in the pathogenesis of AD,PD,and HD.FBXW7 binds to parkin in neurons and ubiquitinates cyclin E1 labeling it for degradation.The excess of cyclin E1 triggers cell cycle activity leading to neuronal apoptosis suggesting a protective role for FBXW7 (Yang et al.,2021).Although not all cases have described a direct correlation with neurodegeneration,APC/C and SCFs ubiquitin E3 ligases target several cell cycle regulators for degradation.SCF ligases act mostly during the G1 and S phase and APC ubiquitinates proteins in G1 and mitosis.In addition,there are cullin-RING E3 ligases that also contribute to proteolytic cell cycle regulation during the G1 and S phases(Zou and Lin,2021).For instance,cyclin D,required for starting the cell cycle,is degraded by APC and several SCF family E3s;degradation of CKIs,such as p21,p27,and p57,needed for transition G1/S,is carried out by SCFSkp2,while if p21 accumulates in prometaphase is degraded by APCCdc20.Not only p21 but also each CKI can be targeted by more than one E3 ubiquitin ligase,so p27 can also be ubiquitinated in the cytoplasm by the Kip1-promoting ubiquitination complex,and p57 by SCFFb112 if degradation is induced by TGFb1 signaling (Zou and Lin,2021).Thus,UPS plays a key role in cell cycle activation and progression,and impairment of this pathway can lead to aberrant cell cycle and apoptosis in neurons.
Role of Cell Cycle Proteins in Neurons
Neurons,muscle cells,and blood cells remain quiescent in phase G0 once they differentiate.The key point for neurons to exit the cell cycle is at the end of the G1 phase and its regulation depends on proteins cdk4,cdk6,cdk2,cyclin D,cyclin E,cyclin A,Rb,E2F,p53,and cdk inhibitors.Thus,increasing the expression of cdk inhibitor p27 promotes G0 entry,and higher expression of cyclin E and D promotes cell cycle progression.However,some of the cell cycle regulatory proteins such as cyclin A1,cyclin D2,Cdk4/6,p21,and p27 are also expressed after neurogenesis,suggesting alternative roles for these proteins in post-mitotic neurons (Akagawa et al.,2021).In addition,although mature neurons maintain their cell cycle arrested in the G0 phase,the cycle is reactivated through Rb phosphorylation by cyclin C and progresses to G1 phase when DNA repair is needed (Joseph et al.,2020).Once DNA is repaired,neurons return to their normal state in the G0 phase of the cycle,but under pathological conditions,neurons do not return to the G0 phase,and instead,an apoptotic pathway is activated,leading to neuronal death.Many factors such as neurotrophic factor deprivation,DNA damage,oxidative stress,impaired proteolysis,and excitotoxicity can contribute to this aberrant cell cycle re-entry.Thus,a decrease of nerve growth factor (NGF) induces cyclin D1 and cell cycle reactivation,amyloid-β-induced cell death appears linked to CDK4/6 and,Rb and inhibitors of G1/S transition avoid cell death induced by DNA damaging agents (Joseph et al.,2020).In addition,many cell cycle regulators including a plethora of cyclins and CDKs undergo changes in their levels in neurodegenerative models reinforcing the idea of cell cycle reentry as a cause of neuronal death (Xia et al.,2019).Increased expression of proliferating cell nuclear antigen (PCNA) has been found in AD and PDin vivoandin vitromodels (Zhao et al.,2021;Lopez-Grueso et al.,2022).Rb is a known inhibitor of G1/S transition through interaction with E2F transcription factors,increases its phosphorylation and releases E2F1 which activates the transcription of cell cycle genes such as cyclin E and A contributing to neurodegeneration (Choi et al.,2019).One of the CDK keys in a neuron’s cell cycle re-entry is Cdk5 since it is connected to all factors that contribute to cell cycle activation in post-mitotic neurons.
Cdk5 and Cell Cycle Activation in Neurons
Cyclin-dependent kinase 5 (Cdk5) is a serine/threonine kinase discovered three decades ago (Pao and Tsai,2021).It was originally thought that cdk5,like other CDks,was aimed to regulate the cell cycle but its main role is related to dendritic growth,axonal guide and formation,and synapse regulation in mature neurons (Allnutt et al.,2020).For this,cdk5 increases with the central nervous system development while most CDKs are decreased (Cortes et al.,2019).Further,unlike the rest of CDKs,cdk5 is activated not only by cyclins,specifically cyclin I,but also by p35 and p39 that are not cyclins.Cdk5,as the rest of CDKs,is expressed ubiquitously,but p35 and p39 are expressed mainly in neurons (Pao and Tsai,2021).p35 can be cleaved by calpain after a neurotoxic stimulus that led to calcium increase into a truncated aberrant coactivator p25 and a p10 fragment (Figure 2).In these conditions,Cdk5 can transform from p35/cdk5 complex with physiological roles during development to p25/cdk5 complex involved in pathological events (Umfress et al.,2022).p25/cdk5 aberrant activity has been implicated in AD,PD,ALS,HD,and neurotoxic insults.
Cdk5-p25 hyperactivation results in ROS accumulation and mitochondrial dysfunction in neurons due to the inactivation of peroxiredoxins I and II.Mitochondrial damage contributes to the increase of ROS and calcium which in turn activates cdk5,so there is a vicious circle that leads to cell death(Figure 2).Inhibition of cdk5 reverses mitochondrial damage,preventing ROS accumulation and neuronal death,which supports the idea that cdk5 acts upstream of these events (Pao and Tsai,2021).Aberrant expression of human p25 in mouse neurons promotes DNA damage by DSBs and increases the expression of cell cycle markers that may lead to cell cycle activation (Pao and Tsai,2021).In addition,Cdk5 is activated by DNA damage and phosphorylates ATM at Ser794 turning on ATM kinase activity and ATM targets p53 and H2Ax(Figure 2).Thus,inhibition of Cdk5-ATM signaling decreases cell cycle reentry induced by DNA damage in neurons and the expression of p53 target genes,inhibiting apoptosis and neuronal death (Kciuk et al.,2022).Cdk5 is related to molecular pathways that induce cell cycle in neurons (Figure 2).Thus,hyperactivation of cdk5 in primary neurons and diabetic mice goes hand in hand with the increase of phosphorylated MAPKs and the induction of neuronal apoptosis and cognitive deficits while these effects were reversed by inhibiting cdk5 (Liu et al.,2019).Cdk5 phosphorylates not only MAPKs but also Rb,STAT3,AR,FAK,AKT,and p21Cip1,activating cell cycle progression (Sharma and Sicinski,2020).Other substrates of cdk5 are Tau and MAP1b which form neurofibrillary tangles,one hallmark of AD (Cortes et al.,2019).Finally,cdk5 is also related to UPS through Cdh1,one of the key cofactors for APC/C in neurons.Phosphorylation of Cdh1 by Cdk5 triggers the disassembling of Cdh1 from APC/C complex,thereby resulting in Cdh1 inactivation (Figure 2).Cdh1 phosphorylation resulting in the inactivation of the APC/C complex enhances neuronal death induced by amyloid β that may contribute to AD (Lapresa et al.,2022).
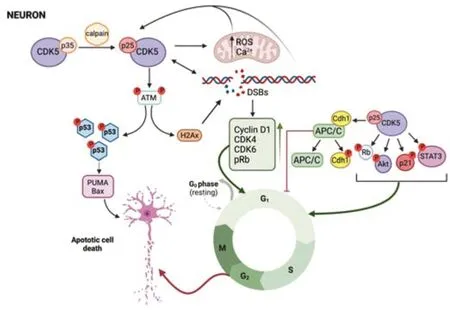
Figure 2|Cdk5 as activator of cell cycle in post-mitotic neurons.
Cdk5 Dysfunction in Neurodegenerative Disorders
Alzheimer´s disease
Alzheimer´s disease (AD) is a progressive neurodegenerative disease characterized by memory loss,impaired cognitive function,behavioral changes,and neuronal death that lead to dementia.The pathological hallmarks of AD are the formation of amyloid β plaques and neurofibrillary tangles (NFTs) (Figure 3A;DeTure and Dickson,2019).The dysregulation of cdk5 is associated with AD pathogenesis,and accumulation of p25 has been observed in Aβ and NFTs,and brain lysates from AD patients (Huang et al.,2020).Cdk5-p25 complex has a much longer half-life than Cdk5-p35 and a different subcellular location.For that,Cdk5-p25 phosphorylates substrates other than physiological ones (Allnutt et al.,2020).One of the main targets of cdk5-p25 complex is tau,a protein that belongs to the microtubule-associated(MAPs).Tau hyperphosphorylation,carried out by both cdk5 and GSK3β,is responsible for its aggregation and NFTs formation (Cortes et al.,2019) and inhibition of cdk5 and GSK3β activity has neuroprotective propertiesin vitroandin vivo(Reinhardt et al.,2019).Cdk5-p25 is also linked to Aβ production,through STAT3-mediated transcription of β-secretase gene (BACE1),cdk5 also phosphorylates amyloid protein precursor (APP) which favors the formation of Aβ (Figure 3A).Mouse models unable to process p35 to p25 showed a decrease of Aβ and plaque deposition while mice overexpressing p25 displayed neuronal death and accumulation of Aβ,the blockade of p25 by crossing both mice reverse this phenotype (Pao and Tsai,2021).In addition,Aβ promotes dysregulation of cdk5 activity since Aβ increases calcium concentration in neurons which activates neuronal calpains,and the cleavage of p35 to p25 exhibits a positive feedback between Aβ and cdk5-p25 complex.Activation of Cdk5-p25 and increased Aβ induce cell cycle re-entry in cortical neurons through hypoxia inducible factor 1alpha (HIF-1α activation that leads to increased expression of cyclin D1,PCNA and DNA synthesis(Figure 3A).Further,inactivation and knockdown of cdk5 using roscovitine or specific siRNA respectively,suppress both HIF-1α expression and cell cycle reentry (Chao et al.,2020).However,there is controversy on the relationship between cell cycle activation by Aβ and the increase of neuronal death since some studies point to a protective role in Aβ toxicity (Ippati et al.,2021).What seems clear is that Aβ activates several regulators of the cell cycle such as CDKs and transcription factors,leading to tau hyperphosphorylation that induces neuronal death (Figure 3A),as has been observedin vitroandin vivousing animal models and in human post-mortem brain samples (Majd et al.,2019;Zhang et al.,2021;Pandey and Vinod,2022).
Parkinson´s disease
Parkinson´s disease (PD) is the second most common neurodegenerative disorder whose symptoms are bradykinesia,tremor,rigidity,myotonia and ataxia.PD is characterized by selective loss of dopaminergic neurons in the substantia nigra pars compacta and cytoplasmic Lewy body (LB) inclusions composed predominantly of α-synuclein (Luo et al.,2022).A wide range of studies have associated Cdk5 dysregulation with neuronal death and PD development (Figure 3B;He et al.,2020;Pao and Tsai,2021;Lopez-Grueso et al.,2022).Cdk5-p25 complex is found to be increased in PD patients’brain,and mouse models and non-human primate model treated with MPTP,a toxin that damage dopaminergic neurons mimicking PD (Park et al.,2019;Pao and Tsai,2021).Conversely,inhibition of cdk5-p25 activity with the inhibitory peptide AAV9-CIP in a MPTP mouse model prevents loss of dopaminergic neurons and alleviates the symptoms in this model (He et al.,2018).Cdk5 can contribute to dopaminergic neurons loss through different ways including oxidative stress and mitochondrial dysfunction (Figure 3B).In fact,several mutations associated with PD occur in proteins related to mitochondria.Cdk5 contributes to mitochondrial damage which impairs fusion and fission processes by phosphorylation of proteins implicated in them.For instance,cdk5 mediates phosphorylation of fission protein Drp1 at S616.Hyperactivation of cdk5 increases Drp1-phosphorylated levels and mitochondrial fragmentation,leading to neuronal death (Park et al.,2019;Rong et al.,2020).Parkin and Pink1,two proteins mutated in familial PD,are also related to maintenance of mitochondrial homeostasis.Pink1 recognizes damaged mitochondria and recruits Parkin that initiates mitophagy,thus,cdk5 phosphorylates Parkin reducing its activity and preventing the initiation of mitophagy (Allnutt et al.,2020).Mitochondrial dysfunction leads to oxidative stress and DNA damage,one of the main pathways to aberrant cell cycle activation (Figure 3B).In fact,several signs of cell cycle activation have been described in PD.Thus,hyperphosphorylated Rb was found in both SN nuclei and hippocampus of PD patients.pRb was also increased in midbrain of rats exposed to MPTP along with the increase of E2F target genes,cyclin D,A,E and B,indicating an activation of cell cycle mediated by cdk5 (Joseph et al.,2020).In addition,cell cycle re-entry is associated directly with proteins mutated in familial PD (Figure 3B).Thus,mutations in Parkin inhibit cyclin E degradation and promote cell cycle progression in familial PD (Gupta et al.,2021),and primary cultures of cortical neurons lacking DJ-1 showed alterations of cdk5 signaling pathway with increase of PCNA,alterations of cytoskeleton proteins,high levels of ROS and α-synuclein,and activation of cell cycle and neuronal death (Lopez-Grueso et al.,2022).In addition,DJ-1 regulates the transcription of Pink1,and the lack of Pink1 has been also related to the increase of neurons entering the S phase of cell cycle and apoptotic cell death in anin vitromodel (Lopez-Grueso et al.,2022).
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive degeneration of motor neurons which produces motor deficits and paralysis.Although most cases of ALS are sporadic,familial cases associated with mutation in genes including TDP43,SOD1,and C9orf72 have been reported(Mejzini et al.,2019).Cdk5 hyperactivation and tau hyperphosphorylation are observed in sporadic and familial patients of ALS (Figure 3C).In different mouse models of ALS,p25 accumulation and cdk5 hyperactivation were also observed,and the severity of the disease in these models was correlated with cdk5 activity.Further,inhibition of cdk5 improved symptoms and increased lifespan (Bk et al.,2019).In addition,several studies indicate activation of cell cycle proteins compatible with cdk5 hyperactivation.Thus,analysis of spinal cord,motor cortex,and sensory cortex from patients with sporadic ALS compared with age-matched controls showed hyperphosphorylated Rb protein and redistribution of E2F1 concomitant with increased levels of cyclin D1,indicating activation of G1 to S that may contribute to motor neuron death (Joseph et al.,2020).In this sense,pRb and E2F1 colocalized with p53,bax,and caspase 3 supporting the idea of increased apoptotic cell death in these diseased neurons (Joseph et al.,2020).
Huntington´s disease
Huntington´s disease (HD) is an autosomal dominant neurodegenerative disease characterized by aggregation of mutant huntingtin (mHtt).Several studies demonstrate that mHtt accumulation induces cell cycle activation(Manickam et al.,2020).In addition,severalin vitroandin vivomodels of HD show an increase in calpain activity and higher p25 levels while cdk5-p35 activity is neuroprotective.Cdk5-p35 complex phosphorylates mHtt at S434 reducing its accumulation and toxicity,cdk5-p35 also phosphorylates mHtt at S1181 and S1201 in response to DNA damage preventing neuronal death associated with p53 (Allnutt et al.,2020).Thus,stimulation of cdk5-p35 activity or inhibition of cdk5-p25 hyperactivation can prevent neuronal loss and HD development (Figure 3D;Pao and Tsai,2021).
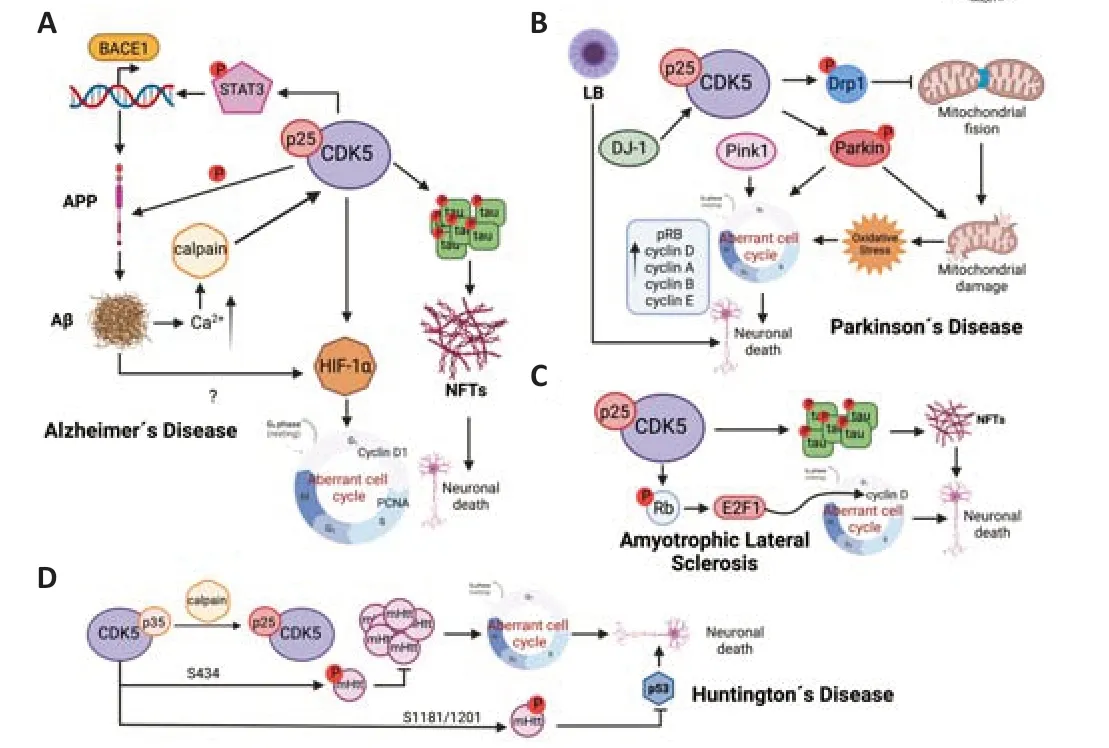
Figure 3|Cdk5 hyperactivation in neurodegenerative diseases.
Conclusions
Aberrant cell cycle re-entry is considered an important mechanism of postmitotic neurons death associated with neurodegenerative diseases including AD,PD,ALS,and HD.Although the exact molecular mechanism behind cell cycle activation in mature neurons is still a matter of intensive research,a growing body of evidence indicates the central role of cdk5 in this event.Hyperactivation of cdk5 seems to be a common feature in neurodegenerative diseases while inhibition of this kinase attenuates disease progression.However,further investigation is needed to clarify the exact mechanism underlying cell cycle activation and neuronal death mediated by cdk5.This helps identify novel therapeutic targets and strategies to specifically target cdk5-p25 complex without affecting other CDKs or cdk5-p35 complex,and thereby open new avenues for the treatment of neurodegenerative disorders.
Acknowledgments:We would like to thank Carmen Alicia Padilla and José Antonio Bárcena and members of BIO-216 group for their support and help in preparing the manuscript.
Author contributions:Manuscript conceptualization and writing: RRA.RRA read and approved the final manuscript.
Conflicts of interest:The author declares no conflict of interest.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Neuro faces of beneficial T cells: essential in brain,impaired in aging and neurological diseases,and activated functionally by neurotransmitters and neuropeptides
- Profiling neuroprotective potential of trehalose in animal models of neurodegenerative diseases:a systematic review
- Recent advancements in noninvasive brain modulation for individuals with autism spectrum disorder
- Vicious cycle of lipid peroxidation and iron accumulation in neurodegeneration
- Cell-based therapeutic strategies for treatment of spinocerebellar ataxias: an update
- Do tau-synaptic long-term depression interactions in the hippocampus play a pivotal role in the progression of Alzheimer’s disease?