
Microglial depletion impairs glial scar formation and aggravates inflammation partly by inhibiting STAT3 phosphorylation in astrocytes after spinal cord injury
2023-01-21 04:42ZhiLaiZhouHuanXieXiaoBoTianHuaLiXuWeiLiShunYaoHuiZhang
中国神经再生研究(英文版) 2023年6期
Zhi-Lai Zhou,Huan Xie,Xiao-Bo Tian,Hua-Li Xu,Wei Li,Shun Yao,Hui Zhang,,
Abstract Astrocytes and microglia play an orchestrated role following spinal cord injury;however,the molecular mechanisms through which microglia regulate astrocytes after spinal cord injury are not yet fully understood.Herein,microglia were pharmacologically depleted and the effects on the astrocytic response were examined.We further explored the potential mechanisms involving the signal transducers and activators of transcription 3 (STAT3) pathway.For in vivo experiments,we constructed a contusion spinal cord injury model in C57BL/6 mice.To deplete microglia,all mice were treated with colony-stimulating factor 1 receptor inhibitor PLX3397,starting 2 weeks prior to surgery until they were sacrificed.Cell proliferation was examined by 5-ethynyl-2-deoxyuridine (EdU)and three pivotal inflammatory cytokines were detected by a specific Bio-Plex ProTM Reagent Kit.Locomotor function,neuroinflammation,astrocyte activation and phosphorylated STAT3 (pSTAT3,a maker of activation of STAT3 signaling) levels were determined.For in vitro experiments,a microglia and astrocyte coculture system was established,and the small molecule STA21,which blocks STAT3 activation,was applied to investigate whether STAT3 signaling is involved in mediating astrocyte proliferation induced by microglia.PLX3397 administration disrupted glial scar formation,increased inflammatory spillover,induced diffuse tissue damage and impaired functional recovery after spinal cord injury.Microglial depletion markedly reduced EdU+ proliferating cells,especially proliferating astrocytes at 7 days after spinal cord injury.RNA sequencing analysis showed that the JAK/STAT3 pathway was downregulated in mice treated with PLX3397.Double immunofluorescence staining confirmed that PLX3397 significantly decreased STAT3 expression in astrocytes.Importantly,in vitro coculture of astrocytes and microglia showed that microglia-induced astrocyte proliferation was abolished by STA21 administration.These findings suggest that microglial depletion impaired astrocyte proliferation and astrocytic scar formation,and induced inflammatory diffusion partly by inhibiting STAT3 phosphorylation in astrocytes following spinal cord injury.
Key Words: astrocytes;coculture;colony-stimulating factor 1 receptor inhibitor;EdU;glia scar;inflammatory response;microglia;phosphorylation;proliferation;spinal cord injury;STAT3
From the Contents
Introduction 1325
Methods 1326
Results 1327
Discussion 1329
Introduction
Spinal cord injury (SCI) is a high-risk trauma that often leads to severe motor and sensory dysfunction (Ramer et al.,2014).Despite the deep understanding of the complex pathophysiology of SCI,few therapeutic strategies are currently available.Inflammatory response,including the activation of resident astrocytes and microglia and infiltration of peripheral blood-derived immune cells,is a vital mediator in determining spinal cord function after SCI(O’Shea et al.,2017;Hellenbrand et al.,2019).
After injury,the lesion core is separated from healthy tissue by an astrocytic scar border,which consists mainly of newly proliferated astrocytes and a small cluster of inflammatory cells,including microglia (Dias et al.,2018).Increasing evidence indicates that inhibiting glial scarring not only does not improve axonal regeneration and functional recovery,but also causes severe inflammatory diffusion and a large lesion cavity after central nervous system(CNS) injury (Anderson et al.,2016;Hesp et al.,2018).Furthermore,studies have demonstrated that astrocyte proliferation and astrocytic scarring play a beneficial role in restricting neuroinflammatory spread and promoting axonal regeneration,ultimately leading to functional recovery (Faulkner et al.,2004;Tsai et al.,2012;Magnusson et al.,2014).Thus,modulation of the astrocytic response and astrocytic scar formation hold great potential for developing strategies for SCI treatment.
Microglia are the first cells to respond to injury,and they modulate the pathological process of injury by changing cell morphology or function,such as enhancing cell secretion and phagocytosis (Davalos et al.,2005;Weerasinghe-Mudiyanselage et al.,2021).However,extensive controversy exists regarding the exact role of microglial activation in neural regeneration and behavioral improvement,particularly when it comes to SCI.Some studies have proposed that activated microglia induce neurotoxic astrocytes and accelerate neural tissue damage and neuronal loss (Kitayama et al.,2011;Yuan et al.,2015;Liddelow et al.,2017;Gerber et al.,2018).Nonetheless,beneficial effects of microglia have also been reported in the context of SCI and contribute to functional recovery.Previous studies reported that loss of microglia disorganized the scar border and aggravated neuroinflammatory expansion and tissue damage (Bellver-Landete et al.,2019;Fu et al.,2020).Experimentally increasing the number of microglia enhanced neurotrophic protein levels and reduced neuronal death (Imai et al.,2007).Overall,more studies are needed to clarify the exact function of microglia in CNS regeneration.In the present study,we advance the understanding of the positive function of microglia in SCI by shifting the focus to how microglia and astrocytes communicate in glial scars.
Given the importance of signal transducer and activator of transcription 3(STAT3) in regulating astrocytic scar formation after CNS injury (Okada et al.,2006;Herrmann et al.,2008),we investigated whether the STAT3 pathway is involved in crosstalk between microglia and astrocytes after SCI.In this study,we depleted microglia by PLX3397 administration in a contusion SCI mouse model and examined the functional recovery and pathological changes.We further explored the underlying mechanism involving STAT3 signaling in the interplay between microglial activation and astrocyte proliferation.
Methods
Animals
A total of 111 adult,female,specific-pathogen-free C57BL/6 mice (aged 8–10 weeks,weighing 20–30 g) were purchased from the Experimental Animal Center of Southern Medical University,Guangzhou,China (license No.SCXK(Yue) 2021-0041) and housed at 23–25°C under a 12-hour light/dark cycle.Mice used for all experiments were naive.No drug tests were done.Five mice were housed per cage.Special attention was paid to reduce the number of animals used and the suffering caused by surgery.This study was approved by the Institutional Animal Care and Use Committee of Guangdong Second Provincial General Hospital,Guangzhou,China (approval No.2019-SB139)on May 7,2021,and experiments were performed in accordance with international laws and National Institutes of Health policies,including the Guide for the Care and Use of Laboratory Animals (8th ed,National Research Council,2011).
Animal injury model and experimental groups
Adult C57BL/6 mice were assigned to three groups by using the random number table method: sham,SCI (SCI only),and PLX3397 (SCI+PLX3397)groups.The experimental groups were subjected to SCI,and the sham group received only laminectomy and the spinal cord was kept intact.After SCI,the PLX3397 group was given PLX3397 by intragastric administration to deplete microglia,and the SCI group was given dimethyl sulfoxide without PLX3397 by intragastric administration at the same time.The sample sizes of the present study were determined in accordance with previous studies (Slomnicki et al.,2020;Nie et al.,2022).The number of mice used was shown inAdditionalTable 1.A full contusion injury was induced with modified forceps as previously described (Liu et al.,2010;Wanner et al.,2013).After mice were anesthetized with 1–3% isoflurane (RWD LIFESCIENCE,Shenzhen,Guangdong,China,Cat# R510-22) mixed with oxygen through inhalation,standard laminectomy was performed and a midline incision was made from T8 to T10.The muscle on each side of the midline was expanded slowly,and the laminae of T9–T10 were removed carefully.Forceps (No.5 Dumont forceps,Fine Science Tools,Foster City,CA,USA) with 0.1-mm tip diameter were passed through the ventral spinal cord.After confirming that both sides of the spinal cord could be compressed,the forceps were fully clamped for 2 seconds.The incision was sutured and mice were placed in the incubator after surgery until fully awake.Buprenorphine was given for the first 3 days post-injury (dpi).Manual bladder expression was performed twice a day after surgery.The mice were perfused at 7,14,or 28 dpi according to the experimental design(Additional Figure 1).
Microglial depletion
PLX3397 (1200 mg/L,Selleck,Shanghai,China,Cat# S7818) was dissolved in 5% dimethyl sulfoxide,1% polysorbate 80,and 0.5% hydroxypropyl methyl cellulose.For the PLX3397 group,PLX3397 was administered at 90 mg/kg per day starting 14 days before surgery and was continued until animals were sacrificed.
5-Ethynyl-2-deoxyuridine injections
To label proliferating cells,200 μg of 5-ethynyl-2-deoxyuridine (EdU;Thermo Fisher Scientific,Waltham,MA,USA,Cat# C10337) in phosphate-buffered saline (PBS) was intraperitoneally injected at 72 hours after surgery.Spinal cord tissue,including the lesion center (1 cm length),were collected at 7 days after surgery and fixed in formalin for 18 hours.Paraffin-embedded spinal cord tissue was sectioned sagittally at a thickness of 4 μm using a microtome(Multicut,Leica Microsystems,Shanghai,China).EdU staining (Salic and Mitchison,2008) was performed using a Click-iTTMEdU imaging kit (Invitrogen,Grand Island,NY,USA,Cat# C10337) according to the manufacturer’s instructions.
Behavioral testing
Basso Mouse Scale (BMS) scoring system was used to assess locomotor functional recovery after surgery (Basso et al.,2006).Briefly,the mice were placed in an open field and two examiners stood on either side of the open field to observe the behavior.Ankle movements,plantar position,weight support,stepping,and coordination were recorded.A score of 0 means completely paralyzed (no ankle movements were observed),and a score of 9 means normal.
RNA sequencing analysis
At 7 dpi,2-mm spinal cord segments,including the lesion center,were collected from different groups (n=3) and homogenized for 2 minutes in lysis buffer.The TRIzol method (Trendel et al.,2018) was used to extract the total RNA.Total RNA was quantified using a Nano Drop and Agilent 2100 bioanalyzer (Thermo Fisher Scientific).Oligo(dT)-attached magnetic beads were used to purify mRNA.After that,mRNA was fragmented into small pieces with fragment buffer to construct an mRNA library.Subsequently,mRNA sequencing was performed on the BGIseq500 platform (BGI,Shenzhen,Guangdong Province,China).Kyoto Encyclopedia of Genes and Genomes(KEGG) pathway enrichment analyses were used to analyze the differentially expressed genes (DEGs).To identify transcripts that were differentially expressed between groups,we defined a criterion of a 1.5-fold or greater difference as DEGs (P< 0.05).Inflammation and fibrosis-related genes were displayed in a heat map.
Measurements of inflammatory cytokines
The levels of tumor necrosis factor-α (TNF-α),interleukin (IL)-6 and IL-1β in the spinal cord were assessed by a Bio-Plex 200 System (Bio-Rad,Hercules,CA,USA,Cat# M6000007NY) according to a previous study (Deng et al.,2021).Briefly,approximately 5-mm spinal cord segments,including the lesion center,were collected and homogenized in lysis buffer.The lysate was transferred to a spin column and centrifuged at maximum speed for 2 minutes at 4°C.Bicinchoninic acid (BCA) assay kit (Beyotime,Shanghai,China,Cat# P0010)was used to quantify the supernatants.Subsequently,assays were performed according the manufacturer’s instructions.Data were analyzed by Bio-Plex Manager software version 6.1 (Bio-Rad,Hercules,CA,USA).
Primary astrocyte culture
Five neonatal,specific-pathogen-free C57/BL6 mice (aged 24–72 hours,body weight 1–3 g) purchased from the Experimental Animal Center of Southern Medical University,Guangzhou,China (license No.SCXK (Yue) 2021-0041)were used to culture primary astrocytes,as previously described with minor modifications (Quintas et al.,2018).Briefly,the whole brain was isolated,cortex tissue was mechanically dissected,and vascular and connective tissue was removed.Then,the brain tissue was minced into small pieces with Dulbecco’s modified Eagle’s medium (DMEM,iCell,Shanghai,China),transferred to a new tube and trypsinized in the incubator (37°C,5% CO2) for 30 minutes.Subsequently,the tissue was centrifuged at 300 ×gfor 5 minutes at room temperature.The supernatant was carefully removed and the pellet was resuspended with 10 mL DMEM.Single cell suspension was obtained by vigorous pipetting.Cell density was adjusted to 1 × 106/mL,and cells were seeded onto a poly-L-ornithine-coated plate (Sigma,St.Louis,MO,USA) and incubated at 37°C in the CO2incubator.The medium was changed every 2–3 days.Mixed glial cells were obtained when cultures were 90% confluent at 14–18 daysin vitro.Mixed cortical cells were purified by gentle shaking at 200 rpm overnight to remove the microglial culture growing on the top layer(Saura,2007).On average,fewer than 5% of microglia were maintained.The design of thein vitroexperiment was shown inAdditional Figure 1.
BV2 cell culture
The immortalized mouse microglial BV2 cell line (Cat# iCell-m011,RRID:CVCL_0182;Blasi et al.,1990) was purchased from iCell Bioscience Inc(Shanghai,China).The culture conditions for microglia were the same as for astrocytes (DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin,incubated at 37°C in the CO2incubator).
Coculture and treatment
To generate a direct coculture system in which cell-cell interactions between astrocytes and microglia were allowed,BV2 cells were seeded together with astrocytes at a ratio of 1:3 (microglia:astrocytes) in a total of 1 mL of DMEM in a confocal dish (NEST,Wuxi,Jiangsu Province,China).To investigate the effects of lipopolysaccharide (LPS)-activated microglia on astrocyte proliferation,LPS(100 ng/mL) was used to stimulate BV2 cells (Kano et al.,2019) for 8 hours before being seeded in the astrocyte-microglia (BV2 cells) coculture system.To assess the role of STAT3 signaling,astrocytes were pretreated with 10 μM STA21 (an inhibitor of STAT3,Selleck,Shanghai,China,Cat# S7951) for 72 hours before coculture.EdU (10 μM) was added to the coculture system to label proliferating cells.The mixed cells were incubated for 24 hours and harvested for immunostaining and western blotting analysis.
Immunocytochemistry and EdU staining
Proliferating astrocytes in coculture were detected by double staining of glial fibrillary acidic protein (GFAP) and EdU.Briefly,mixed cells cultured in a confocal dish were washed twice with PBS,fixed with paraformaldehyde,permeabilized with Triton X-100 (Sigma,Cat# T8787) and blocked with 2.5%goat serum (Boster,Wuhan,China,Cat# AR0009).The primary antibody rabbit anti-GFAP (1:1000,Sigma,Cat# SAB4501162,RRID: AB_10746077) was used to label astrocytes.After 18 hours of incubation at 4°C,the mixed cells were washed three times with PBS and stained with secondary antibody Alexa Fluor 594-conjugated goat anti-rabbit IgG (1:500,Thermo Fisher Scientific,Cat# R37117,RRID: AB_2556545) for 1 hour at 25°C.Click-iT EdU Alexa Fluor 488 assay kit (Thermo Fisher Scientific,Cat# C10337) was used to further label proliferating cells by incubating for 30 minutes at 25°C.After that,the cells were washed thoroughly with PBS,counterstained with 4′,6-diamidino-2-phenylindole (DAPI) mounting media (Sigma,Cat# F6057) and imaged under a confocal microscope (Leica STELLARIS 5,Leica Microsystems).To calculate the percentage of EdU+astrocytes,at least five fields were randomly calculated in each group under 400× magnification.
Western blotting
Western blotting analysis was performed at 7 dpi in accordance with our previous study (Zhou et al.,2020).Three mice per group were sacrificed and 5-mm spinal cord segments,including the lesion center,were quickly dissected.The spinal cord tissue was homogenized in RIPA lysis buffer with 1% protease inhibitor (Millipore,Shanghai,China).Following centrifugation at 15,000 ×gfor 10 minutes,the supernatant was used for further analysis.Protein concentration was subsequently determined by BCA assay (Beyotime),and 50 μg protein from the spinal cord or cell lysates was loaded into a 1.0-mm-thick gel and transferred to the nitrocellulose membrane (Millipore,Cat# IEVH00005),according to standard protocols.The primary antibodies mouse anti-GFAP (1:1000,Santa Cruz Biotechnology,Santa Cruz,CA,USA,Cat# sc-33673),rabbit anti-signal transducers and activators of transcription 3 (STAT3;1:2000,Cell Signaling Technology,Danvers,MA,USA,Cat# 9139),rabbit anti-phospho-STAT3 (p-STAT3;1:2000,Cell Signaling Technology,Cat#9145S),and rabbit anti-β-actin (Sigma,Cat# A5441) were incubated at 4°C overnight.Secondary antibodies peroxidase-conjugated goat anti-mouse IgG(1:5000,Dingguo,Beijing,China,Cat# IH-0031) and peroxidase-conjugated goat anti-rabbit IgG (1:5000,Dinguo,Cat# IH-0011) were incubated at room temperature for 1 hour.The immunoreactive membranes were incubated with enhanced chemiluminescence mix (Thermo Fisher Scientific,Cat#WP20005) for 2 minutes for visualization.Relative protein expression was normalized to β-actin.The band intensities were analyzed using ImageJ software (version 1.8.0,NIH,Bethesda,MD,USA,Schneider et al.,2012).
Immunohistochemistry assays
Animals were perfused with 4% paraformaldehyde at 7,14 and 28 dpi,and the spinal cords were dissected and embedded in paraffin.The samples were sectioned at 4 μm in the sagittal plane.Sections were deparaffinized and rehydrated and then incubated with the primary antibodies mouse anti-GFAP(1:1000,Millipore,Cat# MAB360,RRID: AB_11212597),rabbit anti-GFAP(1:1000,Sigma,Cat# SAB4501162,RRID: AB_10746077),rabbit anti-ionized calcium binding adaptor molecule 1 (Iba1;1:1000,Abcam,Cat# ab153696,RRID: AB_2889406),mouse anti-neuronal nuclei (NeuN;1:1000,Millipore,Cat# MAB377,RRID: AB_2298772),mouse anti-CD68 (1:200,Millipore,Cat#MAB1435),rabbit anti-activated caspase-3 (aCasp3;1:100,Cell Signaling Technology,Cat# 9661,RRID: AB_2341188),mouse anti-Olig2 (1:1000,Millipore,Cat# MABN50,RRID: AB_10807410) and rabbit anti-pStat3 (1:200,Cell Signaling Technology,Cat# 9145,RRID: AB_2491009) at 4°C overnight.For epitope retrieval,the slices were placed in 10 mM sodium citrate buffer and microwaved on high temperature (100 °C) for 20 minutes.Secondary antibodies goat anti-rabbit Alexa Fluor 594 (1:500,Thermo Fisher Scientific,Cat# R37117,RRID: AB_2556545) and goat anti-mouse Alexa Fluor 488 (1:500,Thermo Fisher Scientific,Cat# R37120,RRID: AB_2556548) were incubated at room temperature in the dark for 30 minutes.The slices were mounted with DAPI mounting media (Sigma,F6057).
Image analysis
The stained sections were examined with a fluorescence microscope (Leica Microsystems).We quantified three slices per animal (the midsagittal section and two sections 40 μm lateral to the epicenter) and three fields in each section (lesion center,and caudal and rostral to the lesion epicenter)using ImageJ software.Images were converted to 8-bit and thresholded at a constant setting.The quantification of the lesion area was defined as the GFAP-negative area on sagittal spinal cord sections.GFAP-negative area within 500 μm both caudal and rostral of the lesion epicenter was measured.We manually counted the number of NeuN/aCasp3-positive neurons,EdU/neural marker-positive cells and pSTAT3/GFAP-positive astrocytes at 200× and 400×magnification.
Statistical analysis
All data were expressed as the mean ± standard deviation.Statistical tests were performed using GraphPad Prism version 9.3 (GraphPad Software,San Diego,CA,USA,www.graphpad.com).Normality was examined before we applied any parametric analyses.For data from two groups,an unpairedt-test was used.For BMS score analysis,two-way analysis of variance with Tukey’spost hoctest was applied.For other data from three or four groups,one-way analysis of variance with Tukey’spost hoctest was conducted.Significance level was set atP< 0.05.
Results
Microglial depletion disrupts astrocytic scarring and exacerbates neuroinflammation in injured spinal cord
To assess the potential contribution of microglia to glial scar formation following SCI,we performed a microglial ablation experiment and analyzed its consequences on glial scar formation at 14 days,when the morphology of the astrocytic scar boundary is similar to that at 4 or 8 weeks (Wanner et al.,2013).Immunofluorescence staining showed that in the SCI group,activated astrocytes organized and formed a compact,dense astrocytic scar border.However,when microglia were depleted by PLX3397,GFAP+astrocytes were disorganized around the lesion center,without any particular direction,and failed to form a dense,compact astrocytic scar (Figure 1A).The lesion size was determined by calculating the area of GFAP-negative staining.PLX3397 administration significantly increased the lesion size compared with that of the SCI group (P< 0.0001;Figure 1D).
Next,we investigated the inflammatory infiltrates at 2 weeks postinjury.Activated microglia and peripheral blood-derived macrophages were detected by CD68 immunoreactivity.In the spinal cord of SCI animals,CD68+inflammatory cells were well enclosed in the lesion center by a dense glial scar.However,PLX3397 treatment led to a large number of inflammatory cells spreading into the vicinity across the glial border (Figure 1B).Furthermore,the levels of three critical proinflammatory factors (TNF-α,IL-6,and IL-1β) were detected at 14 days after SCI.Microglial depletion by PLX3397 significantly increased the levels of the three proinflammatory factors compared with the levels in SCI mice (allP< 0.05;Figure 1E–G).Exacerbated inflammatory response often leads to neural cell death after CNS injury.To investigate whether microglial elimination contributes to neuronal death,aCasp3/NeuN colabeling was conducted to examine neuronal death at 14 days after SCI.As shown inFigure 1CandH,aCasp3 expression was very low in sham mice,and the percentage of aCasp3+/NeuN+cells in the PLX3397 group was significantly higher than that in the SCI group (P=0.007).
Microglial depletion inhibits astrocyte proliferation and impairs functional recovery
To detect proliferating cells in the lesion area,we intraperitoneally injected EdU (5 mg/kg of body weight) 72 hours after injury to label the newly formed cells at 7 dpi.Microglial elimination significantly decreased the number of EdU+proliferating cells in the lesion center (Additional Figure 2AandB).Double staining of EdU with GFAP,NeuN,Olig2 and Iba1 was performed to further specify the EdU incorporation in astrocytes,neurons,oligodendrocytes and microglia/macrophages,respectively.EdU was mainly colocalized with GFAP+signals and Iba1+signals,and no NeuN+/EdU+neurons were detected(Figure 2AandB).A marked reduction of GFAP+/EdU+cells was detected in PLX3397-treated mice compared with the SCI mice,suggesting a reduction in the number of newly proliferated astrocytes.
BMS locomotor rating scores were used to assess the behavioral recovery of mice every 3–4 days for 28 dpi.After SCI,BMS scores in both groups were 0 and then gradually recovered.However,the recovery rate of the SCI group was faster than that of the PLX3397 group (Figure 2C).BMS scores in the PLX3397 group were lower than those in the SCI group,and the difference in scores was statistically significant starting at 7 dpi and continuing throughout the observation period (P< 0.01).At 28 dpi,occasional plantar stepping was observed in SCI mice,but only dorsal stepping was observed in PLX3397-treated mice.
Microglial depletion is associated with widespread transcriptional modification in injured spinal tissue
To explore the mechanism of microglial elimination by PLX3397 administration at the transcriptomic level,we next performed RNA sequencing (RNAseq) analysis of triplicate spinal cord samples at 7 dpi.We collected 2-mm segments,including the lesion center,from sham,SCI and PLX3397-treated animals.Total RNA was isolated and further processed for RNA-seq analysis.Analysis identified 1079 DEGs between the SCI mice and PLX3397-treated mice,828 DEGs between the SCI mice and the sham mice,and 656 DEGs between the sham mice and PLX3397-treated mice.KEGG analysis showed that the genes upregulated in the SCI group compared with sham group were enriched in biological processes associated with inflammation and fibrosis (Figure 3A).KEGG enrichment analysis between the SCI group and PLX3397 group showed that PLX3397 treatment suppressed the biological processes related to inflammation and fibrosis (Figure 3B).Using a heat map,we identified genes that were upregulated after SCI and reversed by PLX3397 treatment (Figure 3C).Similarly,gene ontology analysis showed that DEGs between the sham and SCI groups were mainly enriched in categories such as immune system process,inflammatory response,cell adhesion and extracellular matrix organization.Interestingly,we found that many DEGs downregulated by PLX3397 treatment compared with the SCI group were also enriched in cell adhesion,microglia activation,wound healing and extracellular matrix organization (Additional Figure 3).
Microglial depletion inhibited pSTAT3 in astrocytes
Our western blotting data showed that total STAT3 was expressed similarly between the three groups.Phosphorylated STAT3 levels were significantly increased in mice at 7 dpi compared with those in the sham group (P=0.0004;Figure 4AandB).PLX3397 administration abolished SCI-induced pSTAT3 upregulation.Double immunolabeling was used to detect pSTAT3 expression in different cells,and the results showed that pSTAT3 signals were predominantly colocalized with GFAP (Figure 4C).No pSTAT3 signal was observed in Iba1+macrophages/microglia,Olig2+oligodendrocyte cells or NeuN+neurons (Figure 4C).Moreover,the percentage of pSTAT3-positive astrocytes in the spinal cord of SCI animals was significantly higher than that in PLX3397-treated animals (P=0.0002;Figure 4D).
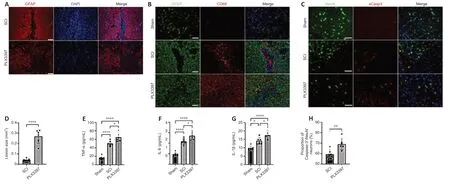
Figure 1|PLX3397 attenuates astrocytic scar formation and increases the inflammatory response following SCI.
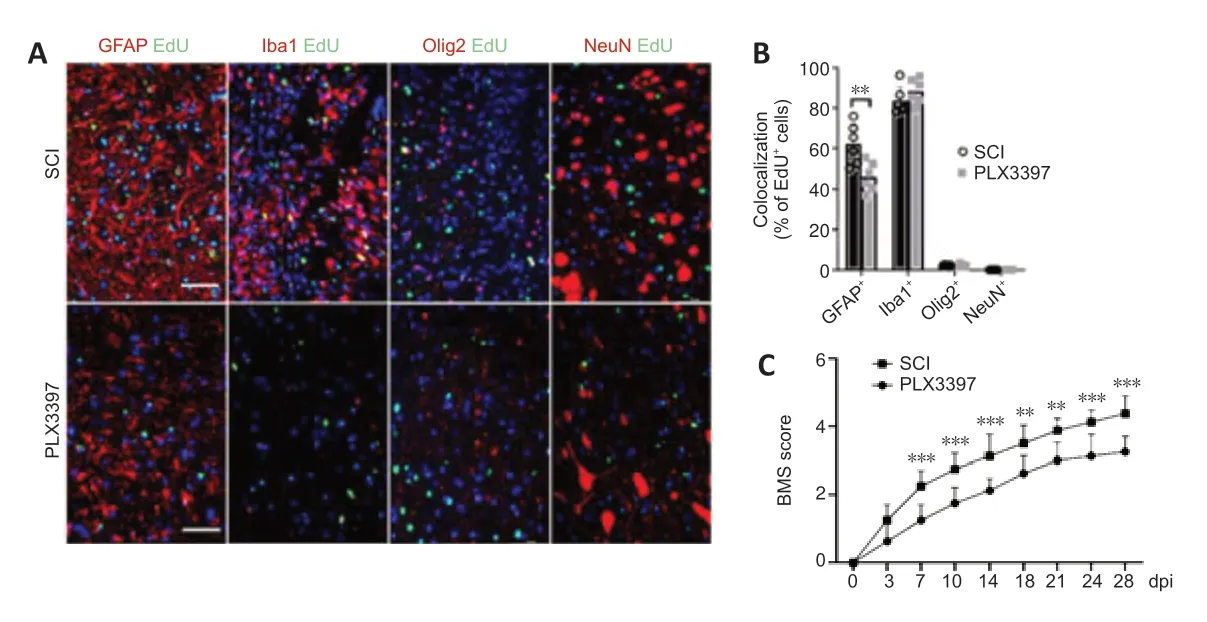
Figure 2|Microglial depletion results in a reduced number of proliferating cells (especially astrocytes) in the spinal cord lesion area.
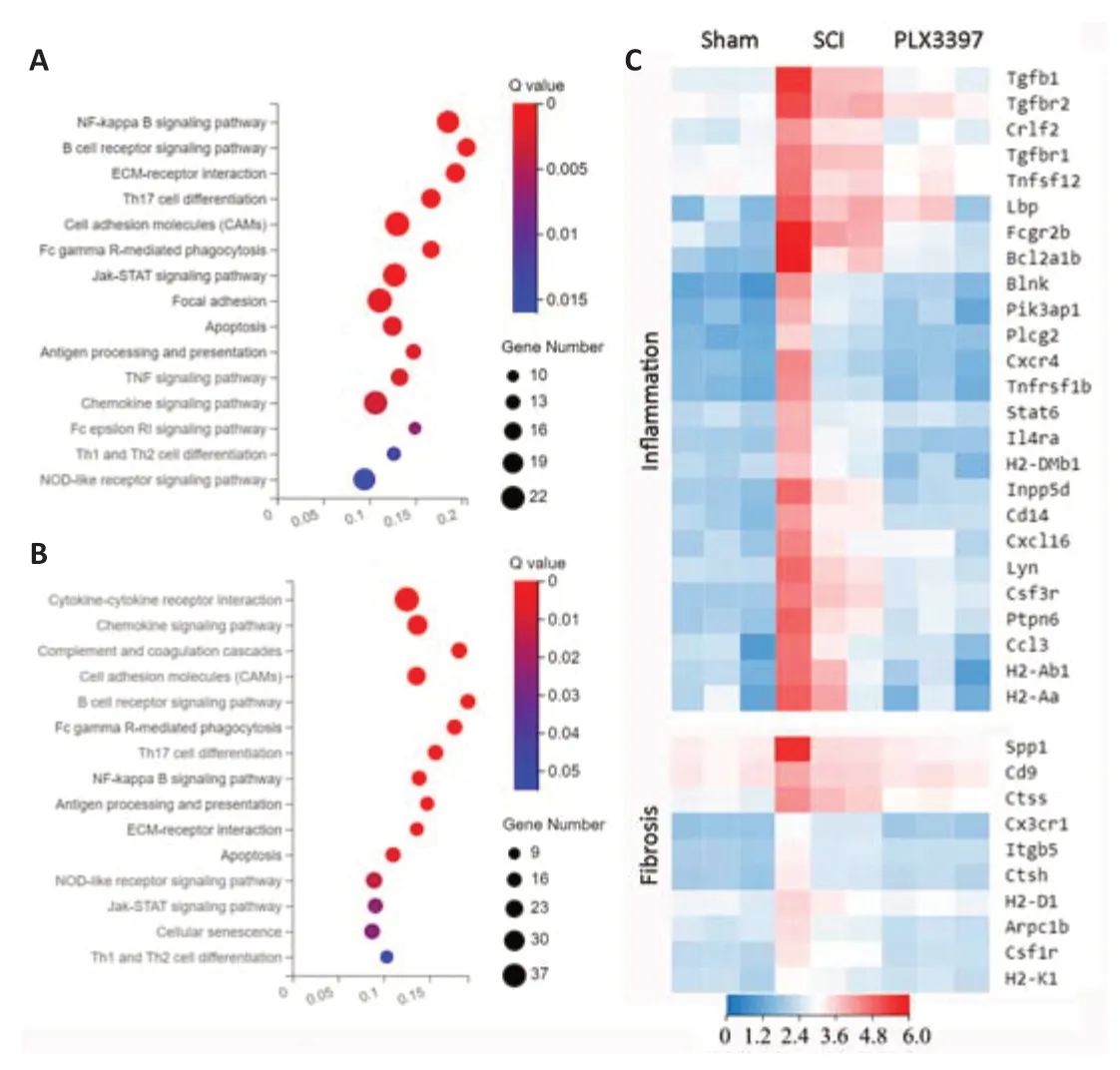
Figure 3|Microglial depletion modulates the transcriptome of spinal cordtissue (RNA sequencing analysis).
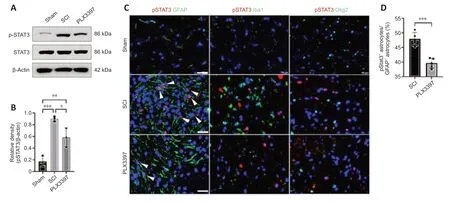
Figure 4|Microglial depletion inhibits phosphorylated STAT3 (pSTAT3) in astrocytes in spinal cord tissue.
STA21 abolishes astrocyte proliferation induced by activated microglia in vitro
To obtain direct evidence that STAT3 signaling activation mediates crosstalk between microglia and astrocytes,we cocultured microglia and astrocytesin vitro.LPS was used to activate microglia (BV2 cells),and STA21 (a STAT3-selective inhibitor) was used to block STAT3 signaling.Western blotting showed that the GFAP level in astrocytes cocultured with LPS-activated microglia was significantly increased by 3.02-fold compared with that in cells cocultured with nonactivated microglia;however,when astrocytes were pretreated with STA21,the activated microglia-induced enhancement of GFAP expression was blunted (Figure 5BandD).Interestingly,we found STA21 only inhibited LPS-activated microglia-induced GFAP expression.Similarly,regardless of whether astrocytes were pretreated with STA21,pSTAT3 expression was low when astrocytes were cocultured with microglia without LPS activation.LPS-activated microglia had markedly increased pSTAT3 level compared with nonactivated microglia,and this enhancement was significantly reduced by STA21 (Figure 5BandC).Next,we examined astrocyte proliferation by double staining with GFAP and EdU,and the results showed that coculture with LPS-activated microglia significantly increased the number of proliferating astrocytes compared with that in the group cocultured with nonactivated microglia.These enhancement effects were inhibited by STA21 treatment (Figure 5AandE).These results indicated that activated microglia promote astrocyte proliferation by modulating STAT3 signaling.
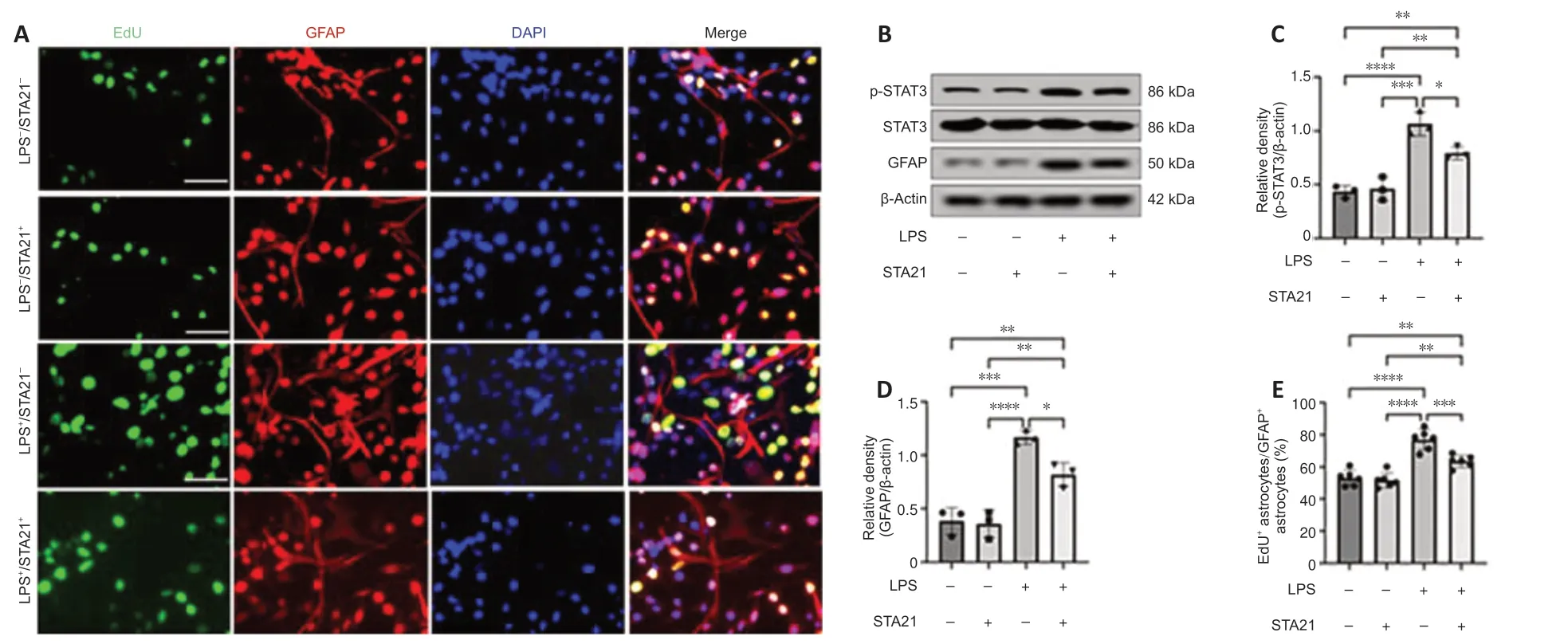
Figure 5|LPS-activated microglia promote astrocyte proliferation in vitro.
Discussion
Our data suggested that microglial depletion led to a reduced number of proliferating astrocytes and disruption of astrocytic scar formation in the lesion area,which were accompanied by an exacerbated neuroinflammatory response and worsened spinal cord function following SCI.Inhibition of the STAT3 signaling pathway in astrocytes was at least partly involved in the regulation of these detrimental effects.
Astrocytic scars consist mainly of reactive astrocytes,immune cells and fibroblasts,which play crucial roles in orchestrating functional recovery after CNS injury (Dias et al.,2018;Hesp et al.,2018;Li et al.,2018;Xie et al.,2020;Yegla et al.,2021).Recent evidence has demonstrated that astrocytic scars aid rather than prevent axonal regeneration after CNS injury.For example,Anderson et al.(2016) used different genetic manipulations to prevent astrocyte scar formation after SCI,and their results indicated that when astrocyte scarring was inhibited,axonal regeneration was no longer observed.Williamson et al.(2021) reported that activated astrocytes played an important role in maintaining vascular integrity in mice with stroke;loss of astrocytes impeded extracellular matrix deposition,leading to a flood of harmful cytokines across the damaged vessel wall into the brain parenchyma,which impaired functional recovery.These processes indicate that reactive astrocytes are a critical element of the glial scar and are essential for promoting functional recovery following CNS injury.The reason for the slow recovery of motor function in PLX339-treated mice is probably related to spontaneous axonal regeneration.Although microglial depletion by PLX3397 administration impaired astrocyte proliferation in the present study,other cells,such as oligodendrocytes and ependymal cells,may still contribute to spinal cord repair.
Microglia are resident immune cells of the CNS that respond rapidly to various stimuli;however,their overall function in CNS pathology remains controversial(Colonna and Butovsky,2017).Traditionally,activated microglia are thought to secrete proinflammatory factors that induce the death of oligodendrocytes and neurons (Liddelow et al.,2017;Li et al.,2021).Nevertheless,recent advances in genetically targeted loss-of-function manipulations and pharmacological reagents to effectively deplete microglia have advanced our knowledge of the specific role of microglia in various experimental contexts.
Microglia elimination resulted in increased neuroinflammation and increased infarct volume after ischemic stroke (Marino Lee et al.,2021).In SCI,the absence of microglia disrupted glial scarring,exacerbated inflammatory cells infiltrates,inhibited neural regeneration,and impeded motor recovery.In contrast,local application of colony-stimulating factor to promote microglial proliferation increased tissue preservation and promoted motor recovery(Bellver-Landete et al.,2019;Fu et al.,2020;Zhou et al.,2020).In an LPSinduced demyelination spinal cord model,microglia proliferated rapidly after injury,dominated the lesion area and restricted neutrophil penetration(Plemel et al.,2020).Similarly,our results showed that microglial depletion disrupted astrocyte proliferation,exacerbated inflammatory infiltration,decreased neuronal survival,and impaired locomotor recovery.In particular,one recent study demonstrated that in neonatal SCI mice,microglia are transiently activated and secrete fibronectin and several peptidase inhibitors to facilitate the connection of injured spinal cord and to resolve inflammation(Li et al.,2020),which further confirms a beneficial effect of microglia in CNS injury recovery.Given that the coordinated interactions between astrocytes and microglia in neuroinflammation resolution are critical for maintaining CNS homeostasis (Damisah et al.,2020),we naturally wondered whether microglia exhibit beneficial effects by mediating the astrocytic response following injury.Interestingly,one study reported that microglia secrete IGF-1,thereby enhancing the number of proliferating astrocytes and promoting glial scarring after SCI (Bellver-Landete et al.,2019).In accordance with this study,we found that microglial depletion induced an overall reduction in EdU+proliferating cells,especially proliferating astrocytes in the lesion area,suggesting that microglia are important for astrocyte proliferation after SCI.
Janus kinase signaling and activator of transcription 3 (JAK-STAT3) signaling is a key regulator controlling astrocyte proliferation and activity (Okada et al.,2006;Tsuda et al.,2011).Interrupted STAT3 pathway activity is sufficient to abolish astrocyte activation and proliferation (Sarafian et al.,2010;Wu et al.,2019;Guo et al.,2021).These advances prompted us to investigate whether the STAT3 pathway participates in mediating the dynamic interactions between microglia and astrocytes after SCI.Our western blotting results demonstrated that the expression of pSTAT3 in PLX3397-treated mice was significantly decreased compared with that in SCI mice.Furthermore,double immunofluorescence assays showed that pSTAT3 staining was predominantly coexpressed with GFAP signaling,and the percentage of pSTAT3+/GFAP+cells was markedly decreased after microglial depletion by PLX3397 compared with that of the SCI group.Taken together,our data suggest that microglial depletion effectively inhibited STAT3 signaling in astrocytes and reduced the number of proliferating astrocytes in the injured spinal cord.
To further investigate whether microglia mediate astrocyte proliferation by STAT3 signaling,a microglia and astrocyte coculture system was used.The results indicated that in the coculture system that lacked pretreatment,astrocytes proliferated slowly.As expected,the number of proliferating astrocytes was significantly increased when astrocytes were cocultured with LPS-activated microglia,and the proliferative effects of LPS-activated microglia were significantly abolished when STA21,an inhibitor of STAT3,was added to the coculture system.Interestingly,we found that STA21 treatment did not abolish all activated microglia-induced astrocyte proliferation,which can be explained by the fact that mechanisms other than STAT3 signaling,including the paracrine effects mentioned above,may also be involved in microglia mediating astrocyte proliferation (Haan et al.,2015;Bellver-Landete et al.,2019).Compared with the coculture system without any pretreatment,STA21 pretreatment did not significantly change the percentage of proliferating astrocytes and GFAP protein level in astrocytes cocultured with untreated microglia,indicating that STA21 did not inhibit astrocyte proliferation in general.These results indicate that inhibition of STAT3 signaling reduced the proliferation of astrocytes induced by LPS-activated microglia.
The present study had several limitations.First,we only investigated astrocyte proliferation at 7 dpi.Further studies to determine how long astrocyte proliferation was affected by microglial depletion will help to clarify whether the effects of microglia on astrocyte proliferation are transient or protracted after SCI.Second,to inhibit STAT3 signalingin vitro,we pretreated astrocytes with STA21 at 10 μM for 72 hours;however,a series of experiments to determine the optimal concentration of STA21 and the duration that STA21 remains effective in astrocytes when it is removed from the medium are warranted to better understand the role of STAT3 in the communication of microglia and astrocytes.
In summary,our study enhanced the understanding of the role of microglia in SCI repair.Our findings suggest that microglia trigger the activation of STAT3 signaling in astrocytes,promote astrocytic scar formation and inhibit the neuroinflammatory response.Strategies to enhance the reparative function of microglia hold great potential for the treatment of SCI in the future.
Author contributions:Study design: HZ,ZLZ;animal model establishment:
HX,XBT,WL;experiment implementation: HX,ZLZ,SY;data analysis: HX,HLX,HZ;manuscript writing: ZLZ,HZ;study supervision: HZ.All authors read and approved the final manuscript.
Conflicts of interest:All authors declare no competing interests.
Availability of data and materials:All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:This is an open access journal,andarticles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:
Additional Figure 1:Timeline of the experiment.
Additional Figure 2:PLX3397 treatment reduced the proliferating cells in spinal cord.
Additional Figure 3:Gene ontology analysis of the differentially expressed genes (DEGs).
Additional Table 1:The number of mice in each experimental group.

- 中国神经再生研究(英文版)的其它文章
- Neuro faces of beneficial T cells: essential in brain,impaired in aging and neurological diseases,and activated functionally by neurotransmitters and neuropeptides
- Profiling neuroprotective potential of trehalose in animal models of neurodegenerative diseases:a systematic review
- Cdk5 and aberrant cell cycle activation at the core of neurodegeneration
- Recent advancements in noninvasive brain modulation for individuals with autism spectrum disorder
- Vicious cycle of lipid peroxidation and iron accumulation in neurodegeneration
- Cell-based therapeutic strategies for treatment of spinocerebellar ataxias: an update