
自然界铀同位素分馏研究进展及展望
2023-01-10 11:38谭凯旋刘振中李春光李咏梅翁宗翔
核化学与放射化学 2022年6期
宋 静,谭凯旋,2,*,刘振中,李春光,李咏梅,翁宗翔
1.南华大学 资源环境与安全工程学院,湖南 衡阳 421001;2.稀有金属矿产开发与废物地质处置技术湖南省重点实验室,湖南 衡阳 421001
铀是天然存在的最重的放射性元素,主要由238U、235U和234U三种天然同位素组成[1]。铀是重要的核材料,其资源开发和理论研究受到国际社会的高度关注。此外,自然界中铀同位素的衰变和分馏在地球科学研究中有重要的理论意义,如利用铀同位素开展宇宙和地质样品的年龄测定[2-3]。早期,由于测定技术的限制,人们普遍认为自然界中238U/235U 不具有分馏效应,近十多年来随着多接收电感耦合等离子体质谱技术的发展和分析精度的不断提高,人们进一步的研究发现铀同位素也和其他过渡金属同位素一样,在特定地质环境下存在同位素分馏作用[4-14]。许多学者[15-35]研究了不同还原条件下的铀同位素分馏特征,发现各种天然材料的238U/235U比率有较大变化,并通过实验研究了238U和235U之间的同位素分馏的可能机制。特别是近年来,一些国外学者[36-49]对地浸采铀等放射性污染区域开展了初步的铀同位素分馏研究,并应用铀同位素研究示踪地下水中放射性核素的迁移转化和环境修复;国内在这方面的研究还较薄弱。本文对近年来铀同位素分馏相关研究成果进行总结,介绍铀同位素分馏研究的新进展以及新应用,指出今后进一步的研究方向。
1 自然界中铀的地球化学行为
铀是主要的放射性元素之一,由原始238U和235U组成,最终衰变为稳定的206Pb和207Pb,半衰期分别为4 468 Ma和703.8 Ma[1]。20世纪初,科学家[50]发现在238U系衰变链中,234U是连续形成的,是铀系衰变链中所有中间核素中寿命最长的子核素。由于铀的难溶性和亲石性,在地球的分异过程中,大陆地壳聚集了约30%的铀,平均含量约为1.3×10-6(质量分数),人们已发现200多种含铀矿物[51]。

根据Dunk等[53]对海洋中铀循环的定量分析(图1),每年海洋中铀的补给量总共约为(53±17) Mmol,现代大洋中铀的总含量达到约19×1012mol;现代大洋中的铀主要有3种来源,分别为陆源碎屑物质、海底地下水和大气尘埃,其中陆源碎屑物质是通过河流运输到海洋中。最主要的铀汇是缺氧的大陆架沉积物、洋中脊的生物成因碳酸盐岩和热液蚀变玄武岩,次要铀汇是含金属沉积物(如铁锰结核)、深海黏土和蛋白石硅石等。由于铀长时间滞留在海洋中和其对氧化还原环境变化的敏感性,特别是沉积环境和全球海洋氧化还原条件的变化对富有机质泥岩中的铀同位素组成有明显影响,铀及其同位素组成已被广泛用作新的全球海洋氧化还原环境演化的示踪剂[18]。应用海洋沉积记录的δ238U值(δ238U为研究样品同位素组成相对于标准物质的千分偏差,‰),对于重建过去局部甚至全球范围内的环境变化具有重要作用。

方框大小代表铀在各地质体中的比例,虚线代表误差,单位:Mmol/a图1 现代海洋中的铀循环[53]Fig.1 Uranium cycle in the modern ocean[53]
2 铀同位素的分析方法
地球化学和地质年代学的发展与元素和同位素丰度测量技术的发展密切相关。
在20世纪30年代,Aston[55]利用质谱检测了挥发性UF6中的238U,得出其丰度大于总铀的97%,这是人类首次用质谱直接测定铀同位素。Nier[12]利用热电离质谱(TIMS)技术首次进行238U/235U原子比值测定,得到铀矿石的238U/235U原子比值为138.9±1.4。在接下来的几十年里,利用气相质谱和UF6测定了铀矿样品的238U/235U原子比值,精度达到±0.2‰水平,这比TIMS测量精度高一个数量级[6]。但气相质谱精度已不足以量化观察到的变化,之后采用“Lunatic I TIMS”仪器测定238U/235U原子比值,只需要用纳克级的样本量,比气相质谱测定的样本量要求低一个数量级,该方法可应用于宇宙化学中[13]。质谱技术的进一步改进,多接收器热电离同位素质谱仪(MC-TIMS)和多收集器等离子体质谱(MC-ICPMS)技术的同步收集能力增强,测定238U/235U原子比值的分析精度提高了一个数量级以上,MC-ICPMS方法对238U/235U原子比值的测量精度达到0.05‰[21]。特别是236U-233U双峰和MC-ICPMS等离子体中的高铀电离源可以用<100 ng的样品量在<±0.1‰水平上测定238U/235U原子比值,拓宽了高精度测定238U/235U原子比值的应用前景[33]。现在,许多实验室常规测定238U/235U原子比值的精度达到了误差<±0.1‰水平,也使用法拉第收集器或与离子计数装置耦合的电子倍增器测定234U[2]。近几年,利用混合1010Ω和1011Ω电阻法拉第收集器代替标准阵列1011Ω电阻和MC-ICPMS,这种方法能在更高强度下测定235U,使238U/235U原子比值测量精度达到±0.03‰水平[9]。
早在20世纪80年代,比利时的欧盟标准物质与测试研究所(IRMM)尝试开发IRMM-072系列标准物质用于铀同位素的测试[13]。之后,Richter等[10]利用高纯度236U和233U制备了双稀释剂同位素标准物质IRMM-3636,并利用IRMM-3636对传统的SRM-960标准物质进行重复测定,将238U/235U原子比值从原来的137.88重新标定为137.837。目前大量实验研究使用了来自美国标准物质与技术研究所和IRMM的几种铀同位素标准物质,包括CRM-112a、SRM-960、CRM-129a、CRM-145、SRM-950a、IRMM-184和REIMP-18a等,其中使用最多的标准物质是CRM-112a。表1列出了不同研究者测定的CRM-112a的238U/235U原子比值以及其它标准物质对应的δ238U值。
目前,铀同位素组成的表达方式主要有两种:一是234U/238U活度比;另一个是δ238U值。例如,Campbell[67]将234U/238U活度比值应用到水循环环境示踪,对铀矿区附近水样的234U/238U活度比进行分析来研究铀在环境中的活动性和迁移机制。Weyer等[68]为了研究235U与238U在自然界中的分馏特征,以δ238U(‰)来表示所研究样品的238U/235U原子比值(r)相对于标准物质该比值的千分偏差,表达式如下:

表1 铀同位素标准物质的238U/235U原子比值和Δ测定值Table 1 Measured values of 238U/235U and Δ of a series of uranium isotope reference
(1)
而同位素分馏系数α和富集系数ε的一般表达式[48]为:
(2)
ε=1 000‰(α-1)
(3)
3 自然界中铀同位素分馏机理与特征
3.1 自然界中铀同位素分馏机理——核场效应
传统的同位素分馏理论是建立在“不同的同位素具有完全相同的电子能,只是动能部分不同”的质量分馏理论基础上的[24]。实验和理论计算认为,铀同位素分馏与氧、硫等传统稳定同位素的质量分馏不同,是与核场效应有关的非质量分馏。核场效应导致重铀同位素更倾向于富集在还原物相当中,这种理论计算结果与传统质量分馏计算结果相反。例如,Bigeleisen和Nomura等[25-26]发现了U(Ⅵ)-U(Ⅳ)交换反应中238U/235U分离异常,该“异常质量效应”与原子核形状和大小差异有关,这是由于电子密度与原子核正电荷占据的空间体积重叠引起的核场效应,能降低原子核周围电子的结合能。之后Schauble[27]通过系统计算,证实了核场效应对Hg等重金属同位素的分馏至关重要,质量效应引起的同位素分馏仅为0.5‰~1‰,核场效应引起的同位素分馏能达到3‰,并且氧化态重金属离子(Hg2+)比还原态重金属离子(Hg0)更倾向于富集重同位素。因此,可以确定铀同位素分馏与核场效应密切相关。
3.2 自然界中铀同位素分馏特征——氧化还原反应
大量实验研究表明,在不涉及铀氧化还原交换反应时,铀同位素分馏十分有限。Shiel等[28]研究含水层中铀迁移及碳酸氢根对U(Ⅵ)吸附-解吸的影响,测量了地下水中的238U/235U原子比值,虽然铀去除量超过50%,但在地下水中没有测出238U/235U原子比值变化,表明在吸附-解吸过程中几乎没有铀同位素分馏。Chen等[29]研究了水溶液中碳酸盐共沉淀对铀同位素分馏的影响,发现铀同位素分馏的大小应与U(Ⅵ)以Ca2UO2-(CO3)3(aq)形式存在的比例成正比,由此推测碳酸盐岩中铀同位素的变化受海水铀的水相形态变化影响,特别是海水pH、Ca2+和Mg2+浓度变化的影响,但这种影响很小。
在涉及铀氧化还原交换反应的实验研究中均发现了明显的铀同位素分馏现象。例如,Bopp等[30]实验研究发现生物刺激铀的还原沉淀可引起δ238U值变化约1.0‰,并通过分析科罗拉多地浸采铀矿区地下水中的238U/235U原子比值的变化来示踪含水层中的污染物铀的迁移转化规律。Rolison等[31]利用海水和沉积物来研究海洋缺氧环境与铀同位素分馏之间的关系,分别采用瑞利模型和封闭体系平衡同位素分馏模型模拟铀还原实验,得出同位素富集系数ε分别为(-0.63±0.09)‰和(-0.84±0.11)‰,这些ε值在微生物还原铀实验的数据范围内,加上U和H2S分布的显著相关性以及在U(Ⅵ)-U(Ⅳ)的还原过程中,238U优先转移到沉积物中,海水中238U/235U原子比值随着铀浓度的降低而减小,表明微生物介导的U(Ⅵ)-U(Ⅳ)还原是影响黑海中铀同位素分馏的主要机制。Stylo等[22]通过实验研究发现金属还原菌还原U(Ⅵ)引起的δ238U变化约为2.0‰,并在还原相中富集重铀同位素(图2)。Bopp[30]和Shiel等[32]分别测定了地浸采铀矿层地下水中的238U/235U原子比值,实验得出的δ238U分馏变化特征与优先去除较重铀同位素的结果相一致,表明铀是通过含水层中的金属还原菌的还原而发生转化。这些研究均反映了铀同位素变化规律与氧化还原环境变化的良好相关性。同位素组成可成为提供自然界生物驱动的铀同位素分馏的“指纹”,238U/235U原子比值给环境和古氧化还原研究提供了新指标,因此,铀同位素被公认为是自然系统中氧化还原极好的示踪剂。
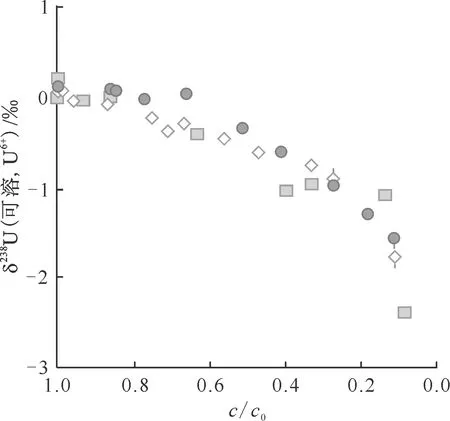
纵坐标为还原菌还原除U后剩余溶液中U的δ238U,横坐标为剩余溶液U(Ⅵ)浓度(c)与初始溶液U(Ⅵ)浓度(c0)的比值◇——无细胞酶提取物,●——碳酸氢盐培养基,■——Widdel低磷酸盐培养基图2 微生物驱动U(Ⅵ)还原和迁移过程中的δ238U变化特征[22]Fig.2 Characteristics of δ238U changes during microbially driven U(Ⅵ) reduction and transport[22]
4 铀同位素组成研究的应用
4.1 铀同位素反演成矿环境
世界上铀矿床产出的地质背景多样,在铀矿床形成的地球化学过程中,氧化还原反应起着重要作用。铀矿床通常可以根据形成时的温度和氧化-还原状态分为两大类型。
(1) 砂岩型铀矿。这类矿床是在接近环境温度、含铀的氧化性地下水向砂岩层运移的条件下形成的。在低温环境中,氧化性流体将U(Ⅵ)配合物从上覆地层中滤出再进入含水层,并向含水层系统深处运移。含有U(Ⅵ)的氧化性流体把水中自由氧耗尽后,在硫化物、还原性微生物或碳氢化合物等还原剂作用下,溶解态U(Ⅵ)被还原为不溶态U(Ⅳ),U(Ⅳ)不断沉淀形成沥青铀矿等[37]。
(2) 岩浆型和热液型铀矿。岩浆型铀矿是分步结晶形成的,导致不相容的铀在高度演化的花岗岩熔体中聚集,直到结晶出富含铀的副相,如晶质铀矿[38]。在太古宙期间,由于大气含氧量低,岩浆型晶质铀矿会在风化过程中保存下来,并积累在河流形成的石英砾岩矿床中[39]。热液型铀矿包括在高温下沉淀形成和岩浆由于烃源岩的沥滤和后期分级结晶而形成的富铀矿物[40]。在火山作用和变质作用过程的高温环境下,热液型铀矿床的沉积机制与砂岩型铀矿床相似,均受U(Ⅵ)-U(Ⅳ)氧化还原作用的控制。当含溶解态U(Ⅵ)物种被还原成不可溶的含U(Ⅳ)矿物时,就会发生铀沉积。这类矿床是最主要的工业型铀矿。
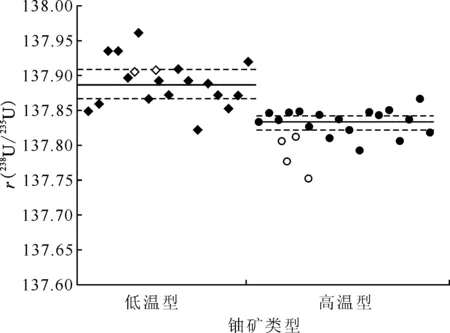
◇,○——数据来自Bopp等[20];◆,●——数据来自Brennecka等[21]图3 不同类型铀矿石的铀同位素组成[20-21]Fig.3 Uranium isotopic characteristics of different types of uranium ores[20-21]
这两类铀矿床,虽然矿床形成温度不同,但成矿过程均与铀的氧化还原状态变化关系密切,因此被统称为与氧化还原作用有关的铀矿床。Bopp等[20]首先研究证实岩浆型和砂岩型铀矿床之间的δ238UREIMP-18a存在约1.0‰的差异,其中砂岩型铀矿床的235U含量明显减少,更富集238U;Brennecka等[21]通过对35个砂岩型和岩浆型铀矿样品的研究发现,所有铀矿石样品的238U/235UCRM-129a原子比值介于137.792~137.961之间,低温型铀矿床的238U/235U 原子比值变化范围很大。虽然Bopp等[20]和Brennecka等[21]的研究中所采用的标准物质不同,但同位素分馏趋势高度一致:形成于低温还原环境的砂岩型铀矿床富集重铀同位素;形成于高温氧化-还原环境的岩浆型-热液型铀矿床富集轻铀同位素(图3),以上两项研究还将238U/235U 原子比值差异与核场效应联系起来,核场效应又与温度有关,因此该趋势可以用核场效应的温度依赖性来解释。Uvarova等[41]通过测定发现不同类型铀矿床的铀矿物δ238U和δ234U值差异较大,认为同位素分馏效应应该反映在铀矿物中铀还原为UO2或化学沉淀形成U(Ⅵ )矿物的238U/235U原子比值上,且随着铀矿物接近矿化,铀同位素组成逐渐改变,与矿床的等级也有关,因此铀同位素可作为勘探媒介,甚至可用于估计矿床大小。
4.2 铀同位素在放射性环境化学与地浸采铀矿山环境修复中的应用
铀矿开采以及铀在能源和武器工业中的应用已经在世界许多地方造成了地下水和土壤的污染。铀具有化学毒性和放射性二重危害,因此铀污染是重要的环境问题。利用传统方法开采铀矿,堆存于矿山附近的含铀废石和尾矿中的铀会随着风化和氧化作用释放到环境中[43];而新型铀地浸采矿工艺是向铀矿区地下含矿含水层注入大量的化学试剂和氧化剂,将不溶性U(Ⅳ)矿物转化为高流动性U(Ⅵ),并且这个转化过程在停采后很长时间依然存在,将给矿山地下水造成严重污染[43,67]。
近年来,少数学者利用U(Ⅵ)-U(Ⅳ)氧化还原过程中同位素分馏变化显著的特点,研究了铀同位素对地浸采区地下水中铀的迁移转化和地下水修复的示踪能力。Basu等[44]研究发现利用还原菌修复后的地浸采铀矿区地下水的δ238U值为-2.49‰~+0.61‰,且下游井的U(Ⅵ)浓度和δ238U值呈降低趋势,矿区的238U/235U富集系数ε约为0.48‰(图4(a));Murphy等[46]研究发现矿化沉积物样品的238U/235UCRM-145原子比值变化非常大,大多数样品的δ238U值介于(-1.30±0.05)‰~(0.55±0.12)‰之间,矿化沉积物沿地下水流动路径逐渐富集238U。而地下水的δ238U值为(-2.39±0.07)‰~(-0.71±0.05)‰,大多数地下水数据显示δ238U值与U浓度有关,U浓度最低的样品的δ238U值最低(图4(b))。这些研究均表明地浸采铀区地下水中污染物铀的降低主要是由还原沉淀引起的。Wang等[23]发现在U(Ⅵ)-U(Ⅳ)的同位素交换反应中,高浓度U(Ⅳ)aq-U(Ⅵ)aq体系比低浓度U(Ⅳ)s-U(Ⅵ)aq体系中铀同位素分馏达到平衡的时间要短,其δ238U值变化为1.64‰,U(Ⅳ)更富集238U。这表明铀同位素分馏变化特征取决于环境条件,铀浓度是关键因素,由此推测铀污染的含水层的铀同位素变化在数年就可能达到平衡,铀同位素组成的变化规律有助于了解地下水修复效果。Brown等[47]探究U(Ⅵ)aq的非生物还原过程发现δ238U值随着铀物种赋存形态的变化而变化。随后Basu等[48]还使用一种新还原菌种来模拟铀还原动力学对铀同位素分馏的影响,发现ε值随着菌种密度和U(Ⅵ)还原速率的增加而降低,测得的ε为(0.36±0.07)‰~(0.96±0.16)‰。因此在解释环境中的铀同位素数据时,应考虑到铀浓度和酶还原的解耦。
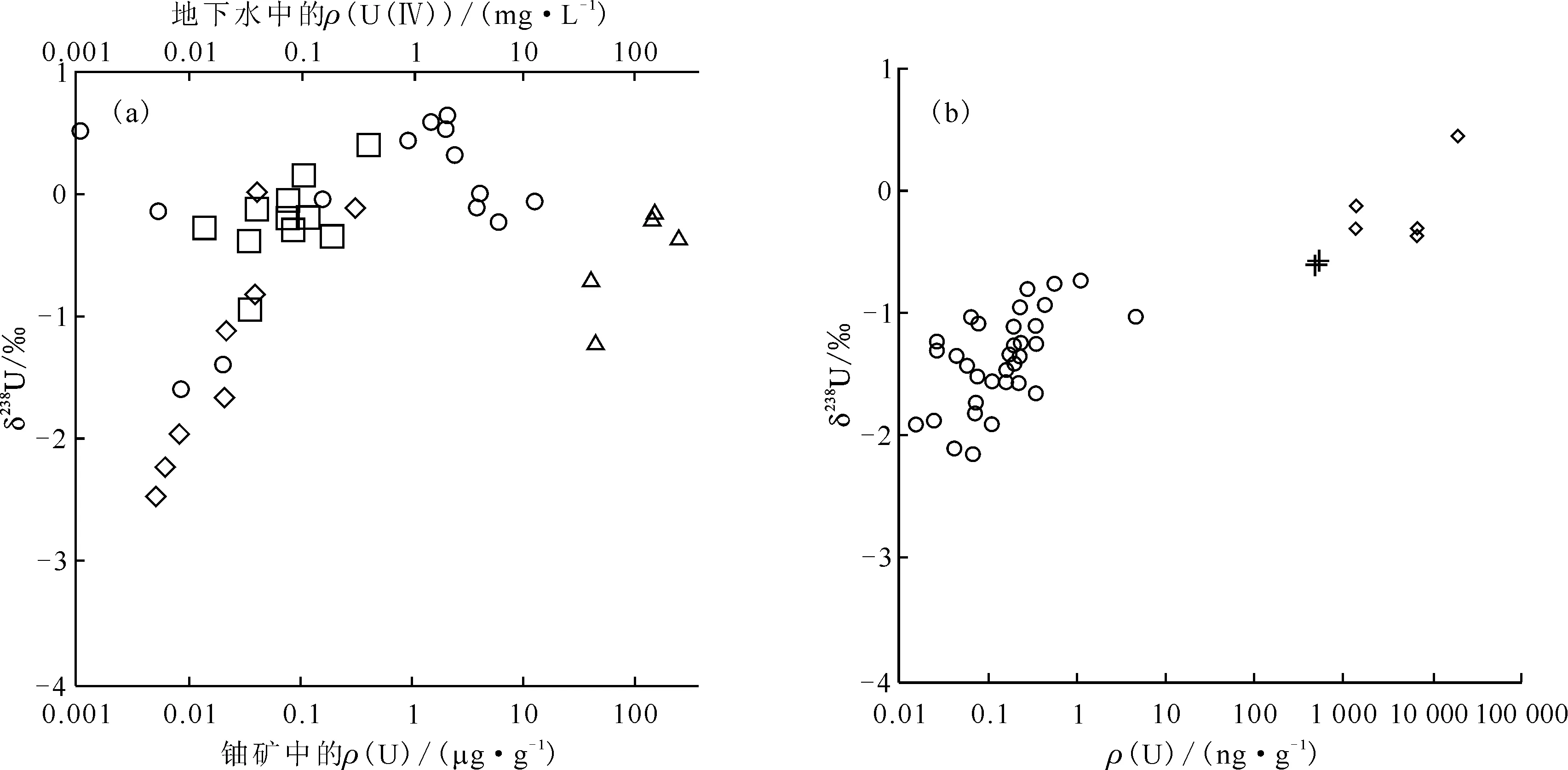
(a)——美国Rosita地浸铀矿[44],□——上游地下水,◇——铀矿下层水,○——下游地下水,△——铀矿沉积物;(b)——澳大利亚Pepegoona铀矿[46],○——砂岩含水层,◇——矿化带沉积物,+——花岗基岩图4 砂岩铀矿地下水和矿化沉积物的δ238UCRM-145与铀浓度的关系[44,46]Fig.4 Relationship between δ238UCRM-145 and uranium concentrations of sandstone uranium mine and mineralized deposits[44,46]
5 总结与展望
在自然界中,铀常以溶解态U(Ⅵ)和不溶态U(Ⅳ)两种形态存在,且主要是由238U、235U和234U三种天然同位素组成,铀同位素组成可以用238U/235U原子比值表示。近百年来,分析测试技术和方法的改进使238U/235U原子比值测定精度已经达到<±0.03‰水平,精度的提高能更好地扩大铀同位素分馏机制的应用前景。铀同位素分馏机理表明核场效应导致238U倾向于富集在还原物相当中,并且铀同位素分馏与氧化-还原环境变化有关,含铀矿物会“记录”不同环境下的铀同位素特征,利用这一原理,通过测定δ238U值,可以为确定铀矿床成因和地浸采铀矿区地下水修复提供依据。铀矿类型主要分成砂岩型、岩浆型和热液型,利用铀同位素反演成矿环境时发现成矿过程均与铀的氧化还原状态变化关系密切,岩浆型和砂岩型铀矿床之间的δ238UREIMP-18a比值差异约为1.0‰,低温型铀矿床的238U/235U原子比值变化范围比高温型矿床大。而在利用铀同位素对地浸采铀矿区地下水中铀的迁移转化进行示踪时发现地浸采铀矿区地下水的δ238U值的变化约为3.0‰,且地下水的δ238U值与U浓度有关,铀的还原沉淀导致地下水δ238U值不断减小。铀同位素在反演成矿环境和放射性环境化学与地浸采铀矿山环境修复方面有广泛应用前景。
迄今为止,实验和模拟研究已经提供了关于驱动铀同位素分馏过程的理论指导。但要深入了解不同铀还原途径中同位素质量平衡限制因素,以及没有铀还原的转移机制中的铀同位素分馏(例如,U-O键配位变化)和动力学铀同位素分馏的影响,在未来的研究中,需要解决的关键问题是:(1) 在近地表环境中,由生物驱动铀还原是否是导致铀同位素分馏的主要因素,生物和非生物还原中铀同位素分馏有何差异,其机理是什么;(2) 铀由于其特殊化学性质和地球化学行为,对研究地质历史演化过程可能比其他同位素具有优势,同时还需要了解地球上导致铀同位素变化的机制;(3) 质量和核体积效应引起的分馏,这两种机制作用方向相反;通过了解这两种机制在不同环境中的作用可能有助于区分潜在的反应途径、同位素平衡或动力学过程的存在,将有助于分析实验中观察到的铀同位素差异,判断是否可以用不同的还原机制来解释,特别是生物还原和非生物还原之间的差异;(4) 生物和非生物还原表现出不同的铀同位素分馏特征,这些特征是否能保留在地质时间尺度上,成为提供生物驱动铀同位素分馏的示踪剂。
铀矿资源在国民经济建设和国家安全方面具有非常重要的经济和社会意义,铀同位素在铀矿成矿过程和在放射性环境化学与环境修复中的研究具有光明的应用前景,可以预见在不久的将来,对铀同位素分馏的研究将进入一个崭新的阶段。
猜你喜欢
河南科技(2021年14期)2021-11-28
矿产勘查(2020年1期)2020-12-28
矿产勘查(2020年11期)2020-12-25
矿产勘查(2020年11期)2020-12-25
矿产勘查(2020年9期)2020-12-25
中国绿色画报(2017年7期)2017-08-02
分析化学(2017年5期)2017-06-21
建材发展导向(2014年6期)2014-11-24
同位素(2014年3期)2014-06-13
同位素(2014年2期)2014-04-16
