
金属卟啉在均相CO2电催化还原中的研究进展
2023-01-09 11:22郁宗华陈宝通刘文博
山东科技大学学报(自然科学版) 2022年6期
郁宗华,陈宝通,刘文博,王 康
(北京科技大学 化学与生物工程学院,北京 100083)
全球碳循环是一个封闭的系统,目前所有化石燃料都来源于数百万年来光合作用固定的CO2。随着人类对能源需求的增加,消耗率与自然固定率之间的不平衡导致了两个主要问题:大气中CO2浓度的上升和可提取化石燃料不可避免的下降[1-2]。过量的CO2排放,导致全球变暖、海平面上升和海洋酸化等严重问题,对人们赖以生存的自然环境造成了巨大的威胁。因此,在全球变暖和能源危机的大背景下,首先要减少对化石能源的依赖,尽可能通过安全、可再生的途径获取化学品,并通过一定的CO2捕集-封存技术[3],在一定程度上缓解大气中CO2的浓度上升问题。其中,利用CO2作为原料并将其转化为有附加值的化学品或燃料被认为是一条有前途和可持续的途径,不仅实现了CO2的利用,而且也解决了能源需求[4-6]。
激活和转化CO2分子的第一步是发生电子的转移(即还原过程)。然而,CO2分子是一个高度稳定的分子,线性的CO2分子和弯曲的自由基阴离子之间存在较大的重组势垒[7]。因此,为了克服CO2还原的热力学和动力学障碍,需要设计高活性的催化剂以稳定中间过渡态。目前,人们提出了基于光催化还原、电催化还原和热催化还原等CO2利用技术[8-9]。其中,电催化技术可以利用可再生能源发电,通过电解装置,将电子传送到电解池中的还原型催化剂,利用电子转移还原CO2。与其他方法相比,CO2电化学转化可以在环境条件下进行,同时具有高度可控的反应步骤和相对较高的转化效率,并且可以由可再生能源驱动/集成到可再生能源系统中,因此在工业规模上更具优势。
催化剂是电催化系统的重要组成部分。在电场作用下,电极表面或者溶液相中的催化剂能够促进CO2分子发生电子转移反应。根据催化剂作用形式的不同,分为均相催化体系和非均相催化体系[10]。非均相催化剂是固定在电极表面的催化剂,能够较快地促进电子转移,提高催化位点的利用率,进而提高催化活性。而且,非均相催化剂的溶解度可以忽略,因此电解液既可以是有机相也可以是水相。此外,非均相催化剂的分离与回收简单[11-12]。但是,非均相催化剂也面临一些挑战:相比于均相催化剂,催化效率低,反应不易控制且催化机理探究困难。而均相催化剂具有确定的分子结构,每个分子性质完全相同;每个催化剂都作为一个活性位点,可以准确评价其反应机理;催化剂溶于反应介质中,分散性好,避免了多相传质问题,催化效率高[13]。不过,均相催化剂也有一些不足。比如,均相体系需要考虑催化剂分子的溶解度。因为有些催化剂只能溶于有机溶剂,如N,N-二甲基甲酰胺(N,N-Dimethylformamide, DMF)和乙腈(acetonitrile, MeCN),不经济且不环保;此外,均相催化剂在使用后不易从反应体系中分离出来,难以回收[14]。但总的来说,均相电催化仍是一种很有吸引力的方法,明确的催化结构使研究者能够建立精确的结构模型,以便更好地分析CO2还原过程中的多质子耦合电子转移过程,从而更好地理解CO2还原机制。目前,研究最多的均相催化剂主要有铑(Rh)、铱(Ir)、钌(Ru)、锇(Os)等贵金属配合物[14-16],以及基于其他过渡金属如Fe、Co、Ni、Zn、Mn等的配合物[17-18]。其中,金属卟啉配合物因其独特的平面分子结构,及其外围基团的可调节性,表现出良好的均相电催化性能[19]。
本研究立足于金属卟啉配合物分子均相电催化剂,简要介绍均相电催化过程中的测试装置、测试方法以及主要评价指标,回顾该领域取得的研究进展,重点对比分析金属卟啉配合物分子催化剂应用于均相电催化还原CO2时的催化性能、效率以及内在的结构依赖机制,增进对该类均相催化剂活性的理解。
1 均相电催化的测试方法和重要评价参数
均相电催化测试中,通常使用的是两室三电极电解池。在评价金属卟啉分子催化剂活性时,需要将不同浓度的金属卟啉溶解在电解液中,测试前用氩气(Ar)或CO2吹扫。测试一般采用循环伏安法(cyclic voltammetry, CV)、线性扫描伏安法(linear sweep voltammetry, LSV)和控制电位电解(controlled potential electrolysis, CPE)等测试方法。涉及的主要参数有:催化过电位η,即催化反应的标准电位与电极电位的差值;转换频率(TOF),表示单位时间内每分子催化剂生成的产物分子数;转换数(TON),表示每分子催化剂生成的产物分子数;法拉第效率(FE),即实际生成物与理论生成物的百分比,可以用来评价催化剂的选择性,数值越接近100%说明选择性越好。电压的单位通常采用一般氢电极(normal hydrogen electrode, NHE)、饱和甘汞电极(saturated calomel electrode, SCE)、银-氯化银电极(Ag/AgCl)和二茂铁电极(Fc/Fc+)等表示。此外,根据log(TOF)~η关系可以得到催化Tafel图,如图1(a)所示,一个性能优异的催化剂,通常具有更低的η,更高的FE、以及更大的TOF0或TOFmax,log(TOF)~η关系曲线位于催化Tafel图的左上方(图1(b))[20]。
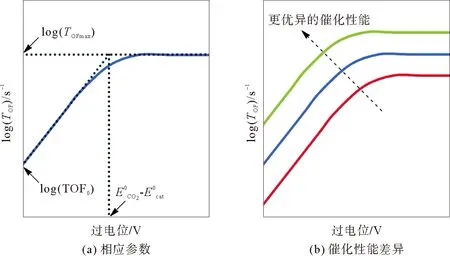
图1 催化Tafel图[20]
2 铁卟啉均相电催化CO2转化研究进展
铁卟啉是最早被应用于均相电催化CO2还原的金属卟啉催化剂。Hammouche等[21]首次探究了包含四苯基铁卟啉(FeTPP)在内的四种铁卟啉催化剂溶解在含0.1 M正四丁基四氟硼酸铵(tetra-n-butylammonium tetrafluoroborate, n-Bu4NBF4)的DMF电解液中的电化学过程,并初步得出了FeTPP催化CO2还原的机理。CV测试发现FeTPP中心的Fe(II)首先被还原为Fe(I),然后随着负电位的增大进一步被还原为Fe(0)。N2和CO2下的CV曲线对比表明:CO2的引入会使Fe(I)/Fe(0)氧化还原峰处出现电流的激增,证明[Fe0TPP]2-中的Fe(0)具有催化CO2还原的活性。基于上述发现,提出了FeTPP电催化CO2还原的可能反应机制,即FeTPP首先被还原为[Fe0TPP]2-,[Fe0TPP]2-经历了一个质子-电子协同转移过程,即[Fe0TPP]2-中心Fe(0)的电子与溶液中的质子转移给吸附的CO2分子,使CO2分子的一个C—O键断裂,随后再经历一次质子和电子转移后,释放出CO。其他3种铁卟啉呈现相同的氧化还原过程,且FeI/0氧化还原峰位出现在更正的电位下,进一步推测卟啉环附近的酰胺(NH—CO)基团的存在可以稳定催化过程中的中间体Fe(I)CO-,提高催化活性。铁卟啉展现出的高效催化CO2还原的活性和选择性也激发了研究人员通过优化铁卟啉衍生物的结构(如取代基的种类、位置等)、催化条件(如电解液的种类、质子添加剂等)以及中心金属的种类等因素,开发更有竞争力的高效催化剂。
2.1 周边取代基团对铁卟啉催化性能的影响
2012年,Costentin等[18]提出在FeTPP的meso位苯环的2号和6号位引入羟基,利用第二配位层提供局部质子源促进催化CO2还原的设想,设计合成了5,10,15,20-四-(2′,6′-二羟基苯基)铁卟啉(FeTDHPP)。为了对比研究,该课题组还合成了无质子源的5,10,15,20-四-(2′,6′-二甲氧基苯基)铁卟啉(FeTDMPP)。以SCE为参比电极,玻碳电极为工作电极条件下,通过CV测试定量比较了1 mM的FeTDHPP、FeTDMPP、FeTPP分别在含1 M四正丁基六氟磷酸铵(tetra-n-butylammonium hexafluorophosphate, n-Bu4NPF6)的DMF电解液中的催化性能。结果表明,FeTDHPP展现出比FeTDMPP更大的催化电流和更小的催化电位。在TOF方面,FeTDHPP催化剂的log(TOF)=3.5,远高于FeTDMPP。此外,FeTDHPP表现出远高于FeTPP+苯酚(PhOH)体系的催化活性,说明FeTDHPP的酚羟基能为催化反应提供较高的局部质子浓度,展示出比简单增加电解液中的酸浓度(PhOH)更好地供质子性能。2014年,Costentin等[22]在FeTDHPP基础上,保留两个相对的苯环上的酚基,并在其余两个meso位苯环上修饰氟原子,设计合成了一种新的铁卟啉基催化剂5,15-二-(2′,6′-二羟基苯基)-10,20-二-(五氟苯基)铁卟啉。相同的实验条件下,新型催化剂的催化电位(-1.55 V,相对于NHE)略高于FeTDHPP(-1.60 V,相对于NHE),而且FCAT表现出更大的催化电流。为了更深入的评价新型催化剂的催化效率,还对比了两种催化剂的TOF与η之间的关系,在η=0.39 V时,FCAT的TOF为240 s-1,高于FeTDHPP在η=0.45 V时的TOF(170 s-1),说明新型催化剂的催化效率优于FeTDHPP,也表明氟原子的吸电子诱导作用可以进一步降低反应的活化能,从而提高催化性能。
除了在铁卟啉的meso位苯环的2/6号位引入羟基,其他质子给体基团(如酰胺基、氨基等)也可以实现局部高质子浓度。为了探究不同氢键基团在CO2电催化还原为CO的动力学调控中的作用,2018年,Sen等[23]以FeTPP为基础,设计合成了铁卟啉FePf、FeEs4和Fe(tBu)4(图2)。以CO2饱和的含有0.1 M四正丁基高氯酸铵(tetrabutylammonium perchlorate, n-Bu4ClO4)和3 M PhOH的MeCN溶液为电解液,对0.5 mM的3种铁卟啉进行了CV测试。测试表明,FePf、Fe(tBu)4和FeEs4的FeI/0电势分别为-1.47、-1.39和-1.35 V(相对于Ag/AgCl),且在FeI/0处均呈现较大的催化电流。然而,波峰分析(foot-of-the wave analysis, FOWA)却显示出FePf的催化速率比Fe(tBu)4和FeEs4快4个数量级。为了深入了解氢键基团所造成的催化速率的巨大差异,通过DFT计算比较了Fe(0)TPP—CO2与Fe(0)Pf—CO2的加合物中Fe—C的距离和∠O—C—O的差别。其中,Fe(0)TPP—CO2中Fe—C键的距离为0.2 nm,∠O—C—O为143°。Fe(0)Pf—CO2中Fe—C键的距离为0.195 nm,∠O—C—O为138.6°,在PhOH存在下,Fe—C键距离则变为0.192 nm,∠O—C—O角度变为135°。基于此,Sen等提出适当的酰胺偶极子的静电作用稳定了中间体Fe(0)Pf—CO2,使CO2上的电子密度增加,进一步使Fe—C的距离缩短,而由于三唑类化合物呈现氢键受体作用,对CO2活化较差,使得Fe(tBu)4和FeEs4展现出的催化活性显著差于FePf。而在引入PhOH后,氢键与静电作用能够共同促进Fe(0)—CO2中间体的活化,提高FePf的催化活性。
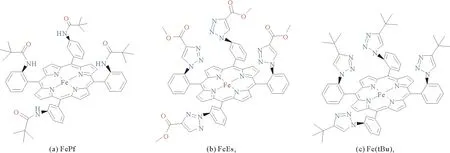
图2 FePf、FeEs4和Fe(tBu)4的结构示意图[23]


图3 FeTPP-Ur和FeTPP-Am分子结构示意图[24]
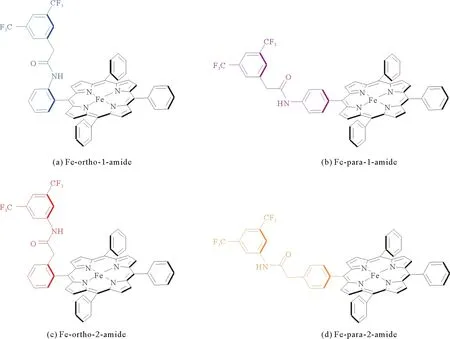
图4 酰胺基化FeTPP的位置异构体[25]
2016年,Ambre等[26]将非质子源型取代基酯基分别引入到FeTPP的meso位苯基的对位、间位和邻位,制备了3种铁卟啉Fe-pE、Fe-mE和Fe-oE,探究酯基的修饰及其位置的改变对CO2还原的催化性能的影响。在CO2饱和的含2 M H2O和0.1 M n-Bu4NPF6的DMF溶液中的CV测试结果表明,Fe-oE在FeI/0处的催化电流最大。通过CPE测试进一步比较了Fe-oE和Fe-mE的催化性能,在-1.66 V (相对于NHE)电压下,Fe-oE的FECO达到98%,远高于Fe-mE的FECO(65%)和FeTPP的FECO(85%),说明Fe-oE具有更高的催化选择性。为了理解催化活性的差异,Ambre等还利用DFT计算了Fe-oE的FeII—COOH和FeI—COOH两种状态的C—O键激活自由能垒(G)。计算表明在FeI—COOH中间体状态下C—O键裂解的势垒更低,并基于此,提出了若体系更富电子,则C—O键裂解的势垒更低,进而表现出更高的催化活性。除了常见的取代基(酚类、脲类、酰胺类、酯基类等),Devi等[27]通过柔性的氧亚甲基连接剂将多个三唑基单元连接到卟啉配体上,合成了含三唑类的铁卟啉催化剂FePEG8T(图5),能够实现CO2到CO的高效还原。以碳纤维纸作为工作电极,在Ar和CO2气氛下,Devi等将0.5 mM FePEG8T在含0.1 M n-Bu4NPF6的MeCN电解液中进行CV测试。如图6(a)所示,与Ar条件下相比,饱和CO2条件下的电流密度仅有少量增加。而当加入2.0 M H2O时,催化电流密度显著增大。这表明FePEG8T催化剂在质子条件下具有较高的CO2还原活性。此外,icat/ip对H2O/D2O浓度的依赖性进一步证明了质子化步骤是催化CO2还原的决速步。在-2.5 V (相对于Fc/Fc+)电位下的CPE结果显示,FePEG8T的催化电流密度达到-17 mA/cm2时,FE达到95%。从Tafel图(图6(b))可以看出,FePEG8T的TOFmax达到5.5×104s-1,在高过电位和低过电位下,FePEG8T催化剂的活性均能与最先进的铁催化剂相媲美。此外,与无铁中心的2HPEG8T和无三唑Fe0T相比,FePEG8T的催化电流密度显著增加,表明了铁中心和三唑单元对催化CO2还原的重要作用。
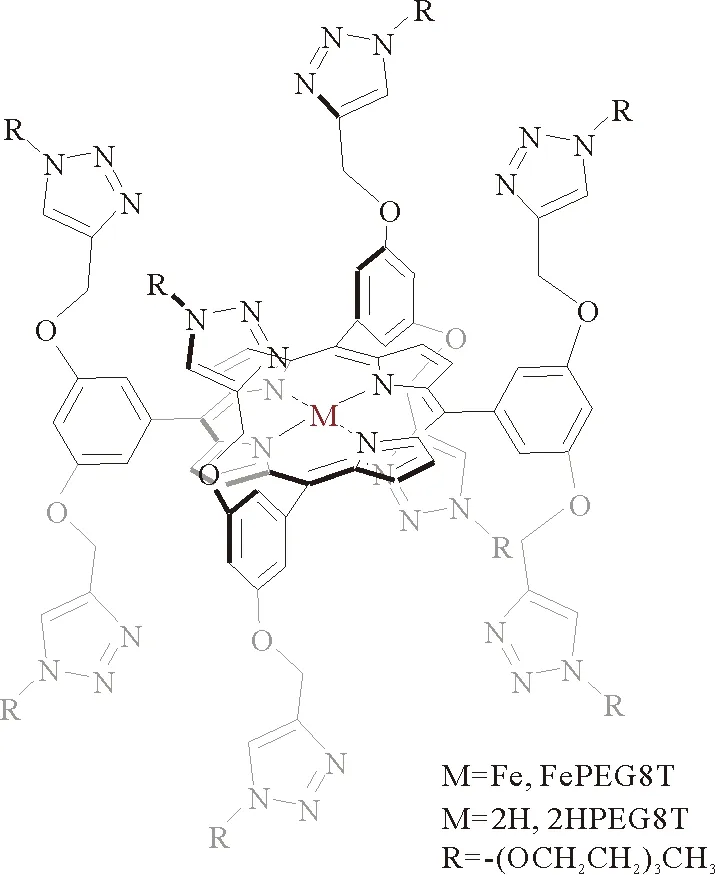
图5 FePEG8T的结构示意图[27]

图6 FePEG8T的电催化活性[27]
大π共轭结构取代基的引入不仅可以改变分子催化剂的电子结构,还可以提供疏水性,有利于在活性中心附近积聚CO2分子。例如Okabe等[28]合成了一系列在meso位有大π共轭基团取代的卟啉,5,10,15,20-四(芘-1-基)铁卟啉(Fe-Py)、5,10,15,20-四(苯基)铁卟啉(FeTPP)和5,10,15,20-四(4-(芘-1-基)苯基)铁卟啉(Fe-PPy)。在含0.1 M n-Bu4ClO4的DMF溶液中,Okabe等对1.0 mM的以上3种铁卟啉配合物进行了CV测试。结果表明,在Ar条件下,FeTPP、Fe-Py与Fe-PPy均表现出可逆的氧化还原波。在CO2气氛下,Fe-Py和Fe-PPy均表现出比FeTPP更大的不可逆阴极电流,说明Fe-Py与Fe-PPy的催化活性高于FeTPP。Okabe等进一步通过CPE测试对比了Fe-Py和FeTPP的催化性能,结果显示,在-2.2 V (相对于Fc/Fc+)电压下,经过1 h的电解反应,Fe-Py和FeTPP的CO析出量分别达到2 978和1 999 μL,对应的TOF值分别为120和60 s-1,对应的TON值分别为4.3×105和2.2×105。表明Fe-Py具有更高的催化反应速率和催化活性,也说明引入π共轭取代基能够提高催化反应性能。
继Okabe等[28]对π共轭取代铁卟啉配合物的探究之后,2019年,Koenig等[29]将苯取代改为噻吩取代,设计了铁(III)四(2-噻吩基)卟啉(FeTThP)和铁(III)四(5-甲基-2-噻吩基)卟啉(FeTThMeP),并与FeTPP的电化学性能进行了对比。首先将1 mM催化剂在含0.1 M n-Bu4NPF6的DMF溶液中进行CV测试。测试结果表明,FeTThP、FeTThMeP和FeTPP的第三可逆氧化还原半波电位分别为-2.02、-2.03和-2.18 V(相对于Fc/Fc+),其中,FeTThP和FeTThMeP的催化电位较FeTPP正移了约150 mV。这意味着FeTThP和FeTThMeP能够在更低的η下进行CO2的催化还原。催化Tafel图进一步说明了噻吩基卟啉能够在较低的η下获得较高的TOF,从而以更少的能量输入实现高效的CO2还原。此外,Koenig等在含0.1 M n-Bu4NPF6和2,2,2-三氟乙醇(TFE)的DMF溶液中,对0.5 mM催化剂进行了5 h的CPE测试,测试结果表明,FeTThP的FECO为(54±1)%,TONCO为17±1,FeTThMeP的FECO为(63±8)%,TONCO=17±3,均与FeTPP的结果相近(FECO为(60±7)%,TONCO为23±2)。该研究说明了噻吩环能够通过扩展分子的π共轭电子结构,使催化剂在较低的过电位下实现与FeTPP相媲美的性能。
2.2 卟啉二聚体结构对催化性能的影响
2015年,Mohamed等[30]从一氧化碳脱氢酶中获得生物灵感,以FeTPP为基准,利用双核配合物的局部推拉机制,合成了铁四苯基卟啉二聚体(o-Fe2DTPP)和1,3-苯基桥联铁四苯基卟啉二聚体(m-Fe2DTPP)(图7)。在含0.1 M n-Bu4NPF6和10% H2O的DMF电解液中的CV测试结果表明,在饱和CO2条件下,o-Fe2DTPP在-1.8 V,相对于NHE下的催化电流(i)与第一还原峰值电流(ip)比值(i/ip)达到200,远大于m-Fe2DTPP和FeTPP的电流响应(i/ip分别为36、35)。说明了o-Fe2DTPP催化活性明显优于m-Fe2DTPP和FeTPP。此外,Mohamed等还在-1.35 V(相对于NHE)的电压下对0.5 mM的o-Fe2DTPP进行了10 h的CPE测试。结果显示出o-Fe2DTPP具有1.15 mA/cm2的平均电流密度,并且10 h内没有下降。计算得出TON和TOF的值分别为1.54×108和4 300 s-1,CO的FE达到95%,说明o-Fe2DTPP表现出良好的催化选择性和稳定性。基于此,文献[30]认为o-Fe2DTPP二聚体高催化活性与其分子内适合的Fe—Fe间分离距离有关。o—Fe2DTPP二聚体中Fe—Fe之间分离范围为0.34~0.40 nm,可以容纳长度为0.23 nm的线性CO2分子。该Fe—Fe分离距离可以使o-Fe2DTPP二聚体中的一个铁中心充当Lewis碱,将电子对推送到配位的CO2分子上,另一个铁中心充当Lewis酸,促进C—O键的裂解并进一步促使CO的形成,二者的协同作用使催化剂呈现出更高的催化活性。而m-Fe2DTPP的分离距离远大于0.40 nm,不利于两个金属间产生协同效应,其催化活性与单金属分子FeTPP相当。
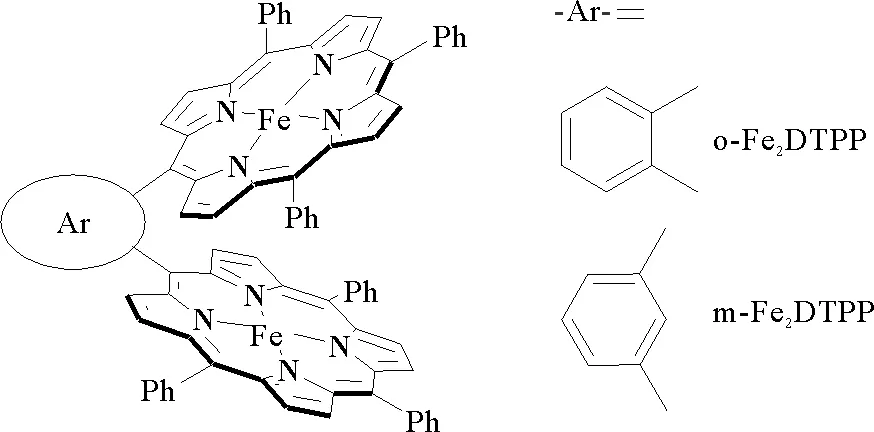
图7 o-Fe2DTPP、m-Fe2DPP的结构示意图[30]
虽然o-Fe2DTPP铁卟啉二聚体呈现出高活性、高选择性和高稳定性,但其过电位仍然较高(η=0.66 V)。为进一步降低过电位,2016年,Zahran等[31]在合成o-Fe2DTPP的基础上,将吸/给电子基团引入卟啉环meso位苯环上,制备6种铁卟啉二聚体化合物,如图8。基于FeTPP的氧化还原机制,将铁卟啉二聚体的Fe中心归因于连续3次的2e-还原/氧化过程,并对0.5 mM的6种铁卟啉二聚体在含0.1 M n-Bu4NPF6和10% H2O的DMF电解液中进行了CV测试。在Ar条件下,FeIFe0氧化还原偶联电位处出现催化电流,表明电化学生成了FeI和Fe0中间状态。在饱和CO2下,这些铁卟啉二聚体在FeIFe0处均表现较大的催化电流(图9(a)),说明6种铁卟啉二聚体均能够促进CO2的电催化还原。从Tafel图(图9(b))中可以看出,这些铁卟啉二聚体均表现出相似的催化性能,但增加铁卟啉二聚体卟啉环上的吸电子取代基,会使催化反应的η降低。例如,全氟苯基(吸电子基团)取代的Fe2DTPFPP的比均三烷(供电子基团)取代的Fe2DTMP的η减小约0.3 V。这些结果表明,铁卟啉二聚体能够通过取代基的改变实现对电子结构的调控,进而高效、低能耗的催化CO2还原。

图8 6种铁卟啉二聚体及其相应单体的结构示意图[31]

图9 在饱和CO2条件下,6种Fe卟啉二聚体的电催化性能[31]
综上所述,FeTPP及其衍生物是均相电催化CO2制备CO过程中具有较高催化活性的催化剂,通过对其取代基种类和位置的改变,能够提高催化CO2还原的活性和选择性。比如,酚基的引入有助于质子转移到中心金属,实现比FeTPP直接添加质子源更高的局部质子浓度,同时能够通过构成氢键稳定催化还原过程中的中间体。氟原子的引入则有效降低了催化η。酰胺键不仅能够利用偶极子的静电作用使CO2上的电子密度增加,还可以作为氢键供体来稳定中间产物。而二聚体则利用局部推拉机制提高了催化剂的性能和稳定性,促进了C—O键的裂解和CO的形成。π共轭取代基的引入则通过扩展π共轭稳定LUMO能级,从而以更少的能量输入实现高效的CO2还原。较大的共轭体系还可以为CO2分子的聚集提供疏水空间,有助于在活性中心附近积聚CO2分子,进而提高催化活性。不同位置的取代基对CO2还原的影响也不同,FeTPP中的邻位取代基显示出更大的催化CO2还原优势,比如,具有邻位酰胺基团的Fe-ortho-1-amide、Fe-ortho-2-amide,以及具有邻位酯基的Fe-oE均较其他位置的催化剂具有更高的催化活性。
2.3 电解液种类对催化性能的影响


2016年,Choi等[36]在探究FeⅢTPPCl催化还原过程中发现,咪唑基离子液体1-丁基-3-甲基咪唑四氟硼酸盐(1-Butyl-3-methylimidazolium tetrafluoroborate,[BMIM]BF4)在催化CO2均相还原制CO过程中能够起到助催化剂的作用。在Ar或CO2条件下,对0.5 mM FeⅢTPPCl在含有TFE和/或[BMIM]BF4的DMF溶液(+0.1 M n-Bu4NPF6)中进行CV测试。Ar条件下的CV表明,与仅加入TFE相比,同时加入TFE和[BMIM]BF4时,FeI/0处的电流显著增加,其电位也从-1.59 V增大到-1.40 V,表明[BMIM]BF4能够促进FeⅢTPPCl的还原。在饱和CO2条件下,仅添加TFE或[BMIM]BF4时催化电流并无明显增加。而同时加入TFE和[BMIM]BF4时,还原电流显著增大,且电位呈现明显正移,FeIII/Ⅱ还原电位较仅加入TFE时正向移动了50 mV,FeI/0电位正向移动了约190 mV。这些结果说明TFE和[BMIM]BF4的结合能够使催化电流增大,并起到降低催化还原电位的作用。
总之,通过改变催化条件也能够获得更有竞争力的催化体系,比如MeCN的加入能够稳定还原中间体,胺的加入则能够增加加合物中CO2的碱度,促进CO2质子化。离子液体中的[BMIM]BF4的咪唑基团能够与催化中间产物产生静电相互作用,使配合物更稳定,从而降低过电位,提高催化活性。
3 其他金属卟啉均相电催化CO2转化研究进展
2019年,Abdinejad等[37]对比了不同数量的氨基取代对Fe、Co卟啉单体催化活性的影响。将0.01 mM的上述卟啉化合物在含0.1 M n-Bu4NPF6的DMF溶液中进行了CV测试。结果显示,具有单取代氨基的Fe、Co卟啉的催化性能最好,且TON和TOF更高,而随氨基数量的增加催化电流会显著降低。基于此,Abdinejad等认为与其他氨基卟啉衍生物相比,单氨基取代卟啉的给/吸电子效应在CO2还原过程中更为平衡,导致其相对较高的催化活性。除了对Fe卟啉和Co卟啉均相催化的研究,2020年,Lashgari等[38]合成了具有柔性三唑单元取代的Zn卟啉配合物ZnPor8T、无中心金属Zn的2HPor8T以及无三唑的ZnPor0T(图10),以碳纤维纸为工作电极,对0.5 mM 催化剂在含2 M H2O和0.1 M n-Bu4NPF6的DMF电解液中进行了CV测试。与Ar条件下相比,CO2条件下的3种催化体系均显示出氧化还原电流的显著增加。而CPE测试结果显示(图11),与无三唑取代的ZnPor0T和无中心金属Zn的2HPor8T相比,ZnPor8T具有更高的电流密度和FECO,在-2.4 V(相对于Fc/Fc+)电位下,电流密度达到6.2 mA/cm2,FECO达到99%,证明了中心金属Zn和三唑单元均能够提高电催化CO2的还原活性。Lashgari课题组[39]还对含有不同数量三唑单元和位置异构的ZnPor8T类似物进行了系统研究,结果显示具有较多三唑单元数量的催化剂具有更高的CO选择性,并揭示了三唑单元能够利用协同作用稳定CO2与催化剂的加合物,进而提高催化效率。在三唑数目的研究基础上,该课题组[39]又探究了构象对锌卟啉的影响,合成了具有两个三唑基的构象异构体(ZnP2T-αα和ZnP2T-αβ),发现取代基的构象不同也会使金属卟啉呈现不同的催化活性。

图10 ZnPor8T、ZnPor0T、2HPor8T分子结构示意图[38]
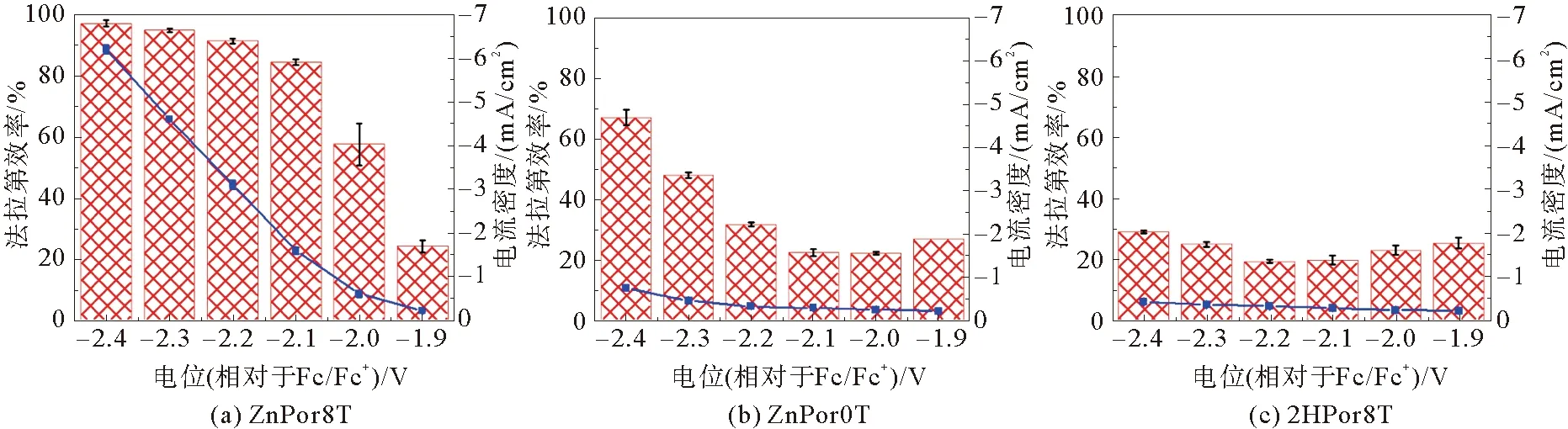
图11 在不同电位下的FECO和电流密度[38]
2020年,Abdinejad等[40]又对镍卟啉进行了研究,合成了两种氮杂环亚胺取代的镍卟啉(Ni-TPP-NItBu和Ni-TPP-adj(NItBu)2),并在饱和CO2条件下对0.01 mM催化剂进行了CV测试。与不带取代基的四苯基镍卟啉(NiTPP)相比,Ni-TPP-NItBu和Ni-TPP-adj(NItBu)2在NiⅠ/0处的催化电流显著增大,说明具有富电子取代基的镍卟啉具有更高的CO2还原活性。此外,-1.1 V(相对于NHE)处的CPE测试表明,与Ni-TPP-NItBu(FECO: 58%,jCO: -1.27 mA/cm2)和NiTPP(FECO: 29%,jCO: -0.73 mA/cm2)相比,Ni-TPP-adj(NItBu)2具有最高的CO选择性(FECO: 62%)和电流密度(jCO: -2.11 mA/cm2)。这些结果说明镍卟啉也能够实现良好的CO2还原催化活性。
4 总结和展望
总结了金属卟啉配合物分子作为CO2还原均相电催化剂的研究进展。其中,铁卟啉是该领域目前研究最为广泛的一类分子,FeTPP及其衍生物的取代基种类和位置的改变,能够显著影响其催化CO2还原的活性和选择性。酚基通过构成氢键稳定催化还原过程中的中间体;给/吸电子取代基的引入有效降低了催化过电位;酰胺键可以作为氢键供体稳定中间产物;二聚体利用局部推拉机制促进了C—O键的裂解和CO的形成;π共轭取代基通过扩展π共轭降低还原过电位,较大的共轭体系还可以为CO2分子的聚集提供疏水空间。不同位置的取代基对CO2还原的影响也不同,铁卟啉中的邻位取代基显示出更大的催化CO2还原的优势。此外,其他过渡金属卟啉(Co、Zn、Ni等)在CO2的转化上的研究虽然还比较少,但是同样展现了良好的应用潜力,在引入合适取代基的基础上,均能实现较好的催化性能。除了催化剂本身,通过改变电解液也能够获得更有竞争力的催化体系,比如MeCN的加入能够稳定还原中间体;胺的加入则能够增加加合物中CO2的碱度,促进CO2质子化;咪唑类离子液体能通过与中间体的静电作用降低过电位,提高催化活性。
目前对金属卟啉均相催化剂活性的认知和调控还存在一些不足之处,比如,吸电子取代基的引入,会导致活性中心的电子密度降低,催化反应虽然能够获得较低的反应η,但是电子密度降低将阻碍质子转移,进而导致TOF降低,反之亦然。又比如,溶剂的种类对催化效率有显著的影响,但是目前溶剂效应对催化活性的影响机制还不清楚。此外,有效利用原位光谱技术捕获催化过程中的活性中间体,结合理论计算,深入研究催化过程中物质转化和电子转移的机理,有助于提供功能导向的分子设计方案。此外,在金属卟啉均相催化剂的研究基础上,还需要进一步拓展研究体系。具有卟啉分子类似的四吡咯大环结构的分子同样有望成为均相CO2还原电催化剂的候选,但目前的研究还很少。比如卟啉的人工合成类似物金属酞菁和萘菁,具有更加稳定的分子结构,并能够提供更强的配体场强度,有望展现出比金属卟啉更加优异的催化活性。总之,深入理解这些电催化剂的静态和动态电子结构及其与CO2分子和还原中间体之间结合等动态过程的催化机制,对开发新型高效的CO2还原催化剂具有非常重要的意义。
猜你喜欢
油气田地面工程(2022年8期)2022-10-02
装备维修技术(2020年5期)2020-11-20
山西农业科学(2020年11期)2020-11-19
矿产综合利用(2020年1期)2020-07-24
化工管理(2020年16期)2020-01-14
火工品(2019年3期)2019-09-02
分析化学(2018年12期)2018-01-22
分析化学(2018年1期)2018-01-18
分析化学(2017年9期)2017-10-16
中小企业管理与科技·下旬刊(2016年12期)2017-01-17
