
水泥熟料预煅烧-烧成工艺特性试验研究
2023-01-06 12:28任强强杨少波
煤炭学报 2022年11期
徐 靖,周 丽,任强强,3,杨少波
(1.中国科学院 工程热物理研究所,北京 100190;2.中国科学院大学,北京 100049;3.中国科学院 洁净能源创新研究院,辽宁 大连 116023)
水泥工业属于传统的煤炭消耗型行业,具有煤炭消耗总量大,单位产品煤炭消耗量高,CO2排放量高的特点。2020年全国水泥熟料总产能18.43亿t[1],生产阶段能耗为2亿t标准煤,碳排放为13.76亿t CO2,分别占全国能源消费总量的5%和全国碳排放的13.5%[2]。GB 16780—2021《水泥单位产品能源消耗限额》要求一级产品的水泥单位产品综合能耗从80 kg/t(标准煤)降低至88 kg/t(标准煤)。在“碳达峰、碳中和”的大背景下,升级传统生产方式、降低生产过程中煤炭消耗量,达成水泥行业节能减排目标的需求迫切。
现有的主流水泥生产技术为新型干法技术,主要设备包括悬浮预热器、分解炉、回转窑和篦冷机。中国建筑材料联合会根据水泥生产技术发展趋势提出第2代新型干法技术的相应要求助力技术创新[3]。从传热效率的角度看,回转窑的热效率最高只能达到60%[4],提升回转窑转速等方法会增加设备的损耗;从反应的角度看,回转窑中承担着固相反应、液相反应和少量未完成的分解反应,复杂的反应融合在一起,并没有达到最佳的反应条件。因此,需要针对回转窑进行改进。已有的改进方式例如薄料快烧的方法[5]和缩短回转窑长度的方法(两档支撑超短窑)[6],通过缩减回转窑内过渡段反应,避免物料活性丧失。中国建筑材料总院采用预烧成的方式,提高分解炉温度至1 100 ℃,在分解炉中短暂停留后进入回转窑。分解炉温度提升后,碳酸钙分解反应在回转窑中进一步减少,证明了提升入窑生料温度有助于固相反应进行[7]。减少回转窑中发生的反应,甚至替换回转窑成为一种趋势。中国科学院工程热物理研究所提出流态化预煅烧-烧成工艺,采用流化床替代回转窑提高传热效率;并将部分固相反应(硅酸二钙形成)与液相反应(主要是硅酸三钙形成)分开,使用煅烧炉和烧成炉取代分解炉和回转窑,使得整个熟料的加热过程分段进行。流态化煅烧时粉末流化特性和颗粒形成机制在之前的工作中论述[8-9]。
笔者对预煅烧-烧成工艺进行研究,通过单矿物合成,确定煅烧过程中的表征矿物;通过主要矿物质量分数变化,确定预煅烧-烧成过程的耦合条件,预煅烧-烧成过程对硅酸三钙形成的影响;并对预煅烧-烧成工艺的理论熟料形成热和减排能力进行计算。
1 试 验
1.1 流态化预煅烧-烧成工艺介绍
预分解窑与流态化预煅烧-烧成工艺简化流程如图1所示,2个流程主要的区别在于回转窑中的固相反应被移出至煅烧炉中进行,且煅烧炉和烧成炉改进为流化床。笔者仅研究加热过程的变化对矿物形成的影响,排除流动过程对矿物形成的影响,故采用水平管式炉进行试验。
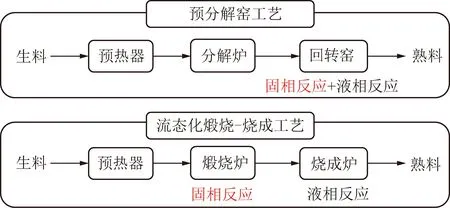
图1 不同工艺流程示意Fig.1 Schematic diagram of different process flow
1.2 试验装置
试验在实验电炉上进行,装置布置如图2所示。2个水平管式炉串联布置,按照试验需求分别设定2个水平管式炉的工作参数。试验时,先将2个水平管式炉分别升温至设定温度,待温度稳定后,将装入样品的瓷舟立即放入水平管式炉1号恒温区内(初始位置),停留设定时间后用送样杆送入2号恒温区(最终位置),停留设定时间后立即取出冷却。冷却后的样品放入干燥箱中干燥备用。水平管式炉1号模拟的是煅烧炉中的加热过程,水平管式炉2号模拟的是烧成炉中的加热过程。
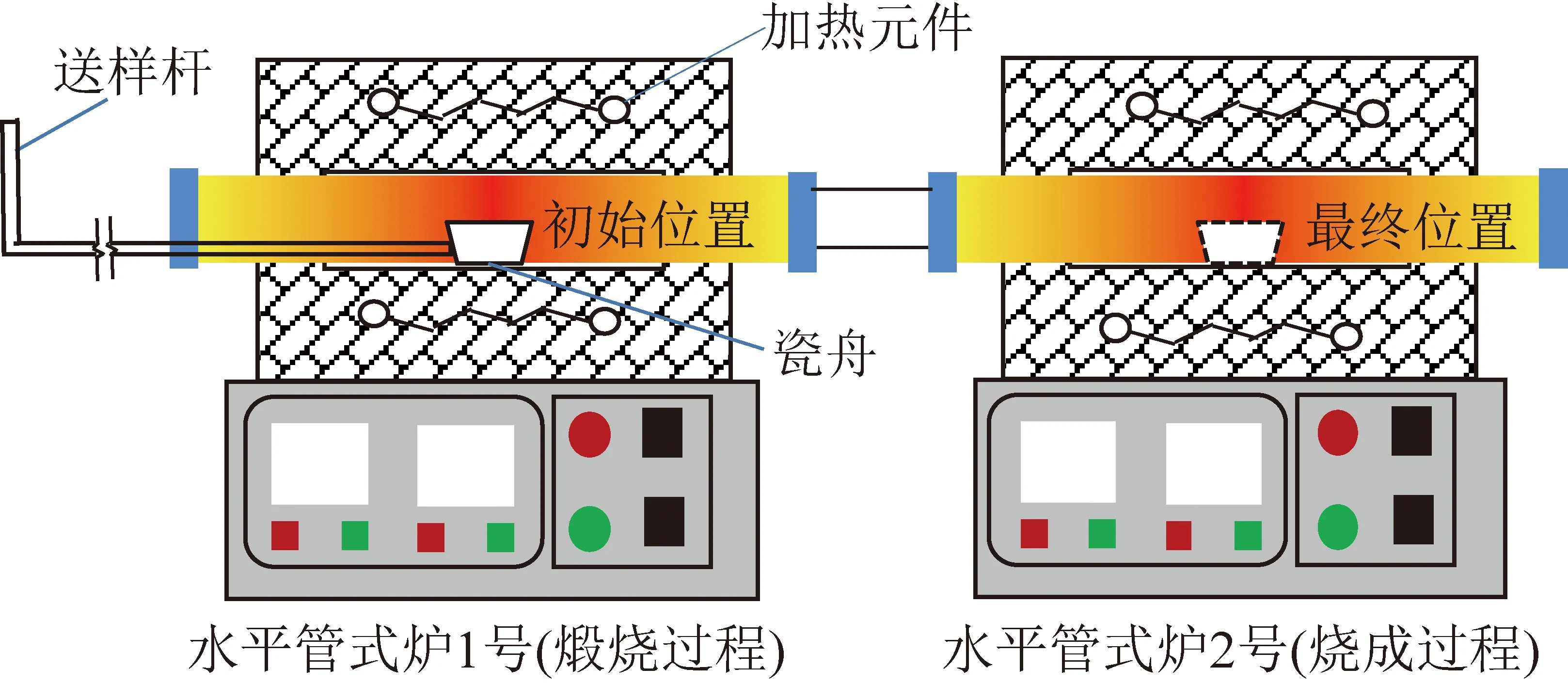
图2 试验装置示意Fig.2 Schematic diagram of the test device
1.3 试验原料
试验原料采用国药集团分析纯化学试剂CaO,SiO2,Al2O3,Fe2O3,生料取自宁夏胜金水泥厂,其组成见表1。按照水泥行业通用表达,水泥熟料主要成分3CaO·SiO2(硅酸三钙)表示为C3S,2CaO·SiO2(硅酸二钙)表示为C2S,3CaO·Al2O3(铝酸三钙)表示为C3A,4CaO·Al2O3·Fe2O3(铁铝酸四钙)表示为C4AF。

表1 水泥生料的组成成分
1.4 测试方法
试样采用XRD分析(Panalytical,Empyrean,荷兰),矿物质量分数采用Rietveld全谱拟合方法计算。矿物形貌采用扫描电镜观察(JEOL,JSF-6700F,日本)。
2 煅烧过程对矿物形成的影响
2.1 单矿物合成研究
为了探究煅烧过程中的矿物形成情况,通过单独合成反应过程中的主要矿物,比较固相反应转化率来确定固相反应的反应情况。熟料的主要成分为C3S,C2S,C3A和C4AF,由于C3S主要在1 300 ℃以上生成,900~1 200 ℃内的生成量极少,主要考虑C2S,C3A和C4AF生成,而C4AF由C3A,CaO与SiO2一同反应生成,故只考虑C2S与C3A。生料中CaO与SiO2的摩尔比通常高于3∶1,在这种摩尔比下,C2S是CaO与SiO2的反应中唯一的产物。只有CaO与SiO2的摩尔比为1∶1甚至是1∶2时才会出现其他的含硅相,例如C3S2(Ca3Si2O7)和CS(CaSiO3)相[10]。CaO与SiO2反应得到的钙硅相产物,按照Ca2+浓度变化排序为CaO→C2S→C3S2→CS→SiO2。同样地,C3A形成和CaO/Al2O3摩尔比有关。CaO过量时倾向于生成C3A,不足时则会生成C12A7,CA。CaO与Al2O3反应按照Ca2+浓度变化排序得到的钙铝相产物的顺序为CaO→C3A→C12A7→C2A→CA→CA2→CA6→Al2O3。试验采用国药集团分析纯试剂CaO,SiO2,Al2O3进行矿物合成。按照CaO∶SiO2摩尔比为2∶1与CaO∶Al2O3摩尔比为3∶1在900~1 200 ℃下煅烧30 min,观察固相反应转化率的变化。固相反应转化率G可以表示为
(1)
其中,m(CaO)为总的CaO质量;mf(CaO)为反应剩余的CaO质量。2种矿物的固相反应转化率都随着温度的升高而升高,如图3所示。

图3 固相反应转化率与煅烧温度的关系Fig.3 Variation of solid-phase reaction conversion withcalcination temperature
C2S的固相反应转化率高于C3A的固相反应转化率,C2S生成要比C3A更快。在固相反应中选用C2S代表固相反应是可行的。生料的液相初析温度在1 250~1 280 ℃,温度从1 200 ℃继续上升会造成液相出现影响流化,而升高温度有助于固相反应进行,故选1 200 ℃作为煅烧温度。
2.2 煅烧时间对矿物形成的影响
预煅烧-烧成工艺的加热时间包括煅烧时间和烧成时间,假定烧成温度为1 350 ℃,烧成时间为30 min时,研究煅烧时间变化对矿物形成的影响,试验原料为宁夏胜金水泥厂生料,结果如图4所示。C2S质量分数和f-CaO(f-CaO表示未反应完的CaO)质量分数随着煅烧时间增加而先下降后上升,C3S质量分数随煅烧时间增加先上升后下降,C2S+C3S质量分数先上升后下降再上升。煅烧时间在10 min以内时,CaO被正常消耗,CaO活性下降还未导致f-CaO质量分数上升;10 min后则因为形成CaO晶体团聚体等原因造成f-CaO质量分数上升。C3S质量分数的变化趋势与f-CaO质量分数的变化趋势正好相反。C2S质量分数先降低而后升高,C2S+C3S质量分数先增加后减少再增加。该现象符合CaO→C2S→C3S的实际过程,由于CaO反应活性的降低,C2S→C3S的过程被抑制,剩余C2S和CaO质量分数高,转化的C3S少。
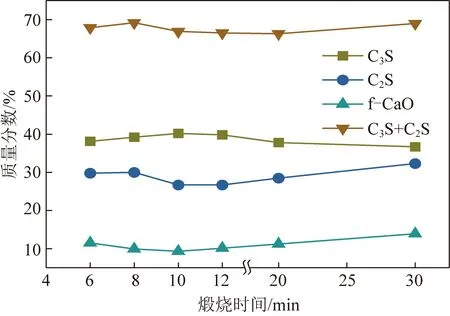
图4 熟料矿物质量分数随煅烧时间的变化Fig.4 Variation of clinker mineral content with calcination time
KAEWWICHIT等[11]在研究微波煅烧水泥熟料时采用相似的煅烧策略,在1 200 ℃采用微波加热的方式分别间隔煅烧生料5~40 min后,将煅烧后的生料放入管式炉中,再在1 300 ℃和1 350 ℃分别煅烧30 min,通过比较7 d抗压强度,发现在1 200 ℃,9 min+1 350 ℃,30 min条件下煅烧,7 d抗压强度最高且接近商业熟料强度。这一结论和图4中的C3S质量分数变化是吻合的,因为7 d水化强度时,C3S的水化强度远大于C2S的水化强度,C2S的水化强度又大于C3A和C4AF的水化强度,因而可以近似认为C3S质量分数最高时,熟料水化强度最高。但仅考虑7 d水化强度是较为片面的,最佳的方式是C3S质量分数高时,C3S+C2S质量分数相应的也高。同时,煅烧时间短时,能量消耗减少,煅烧炉内的停留时间应当在8~10 min。
3 烧成过程矿物形成分析
3.1 烧成温度对矿物形成的影响
试验采用宁夏胜金水泥厂生料,煅烧过程设置为1 200 ℃停留10 min,烧成过程设置为不同的烧成温度停留30 min。不同烧成温度对矿物形成的影响不同,采用Rietveld全谱拟合精修的方法得到的矿物质量分数如图5所示。
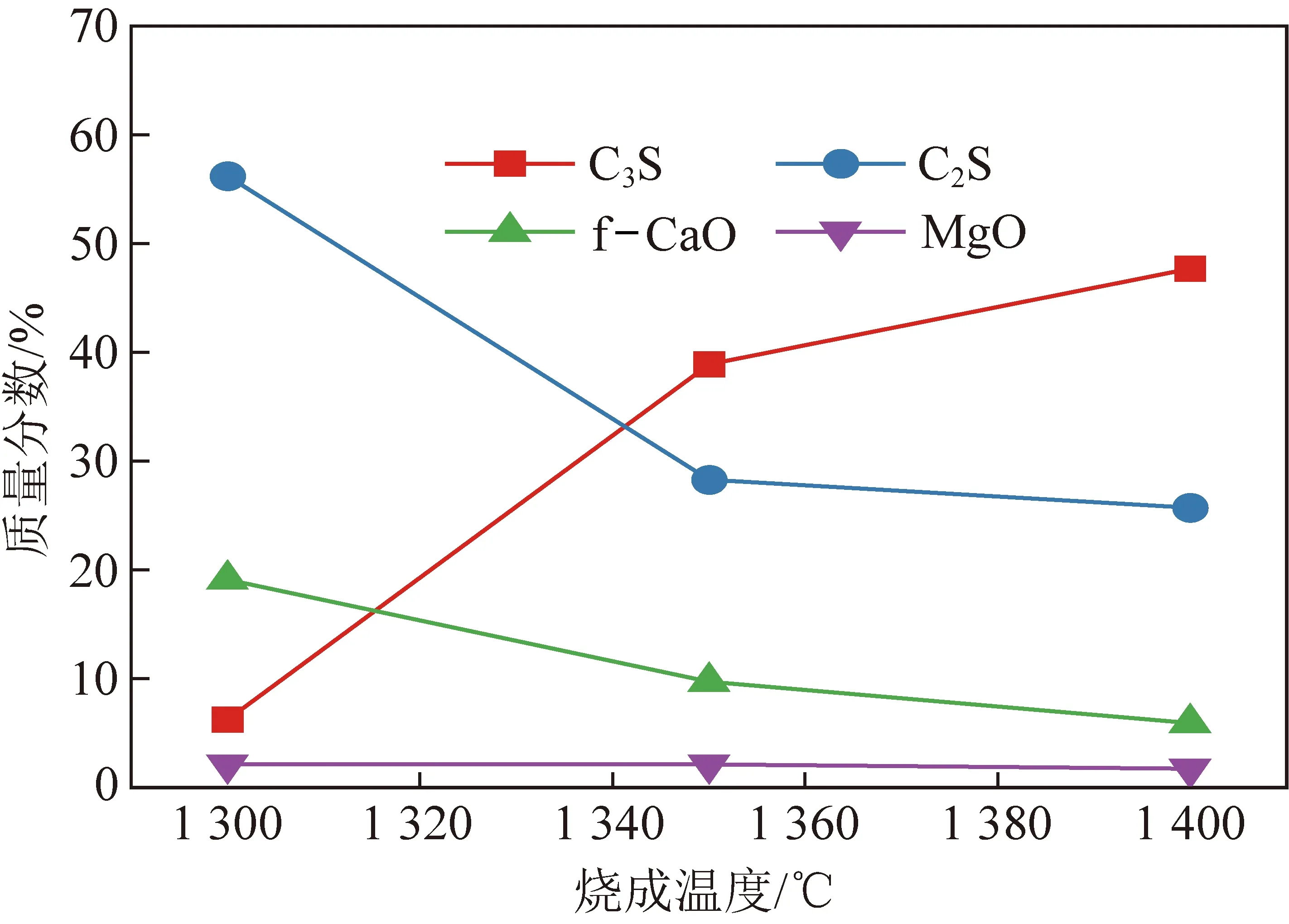
图5 熟料矿物质量分数随烧成温度的变化Fig.5 Variation of clinker mineral content with sintering time
烧成温度升高,C2S质量分数下降,C3S质量分数上升,f-CaO质量分数下降。1 300 ℃ C3S的生成速率为0.206%/min,1 350 ℃时为1.34%/min,1 400 ℃时为1.59%/min。烧成温度从1 350 ℃上升至1 400 ℃时比从1 300 ℃上升至1 350 ℃的增长幅度降低,提升烧成温度对于C3S仍有促进作用,但促进作用减弱。烧成温度升高时,主要的矿物转变过程是C2S与CaO反应转化为C3S。在这过程中,烧成温度一方面影响液相的质量分数,另一方面影响Ca2+的扩散速率[9]。1 400 ℃时熟料中的f-CaO的质量分数为5.9%。根据GB/T 21372—2008的要求,普通硅酸盐水泥基本化学特性中f-CaO的质量分数≤1.5%,1 400 ℃煅烧时得到的熟料,f-CaO的质量分数仍然不能够达标。f-CaO的质量分数较高的原因是熟料的率值不匹配。普通熟料的率值:石灰饱和系数为0.9,硅酸率为2.65,铝氧率为1.45;仅加热生料得到的熟料率:石灰饱和系数为1,硅酸率为3.04,铝氧率为1.27。石灰饱和系数过大时造成CaO质量分数高,烧成难度增加,熟料中剩余的f-CaO质量分数相应增加。采用试剂配制石灰饱和系数为0.9、硅酸率为2.5、铝氧率为1.5的熟料,配制的熟料在1 200 ℃恒温10 min、1 350 ℃恒温30 min后,测量f-CaO质量分数,测量结果为1.4%,基本达到要求。
不同烧成温度下的熟料形貌如图6所示,熟料由数量众多的“小块”黏结而成,1 300 ℃时的熟料表面较为圆润,能够明显地看出烧结的界限。1 400 ℃时的熟料表面不平整,能够观察到熟料表面的晶纹。平行双晶纹或交叉双晶纹是C2S矿物的特征,1 400 ℃下C2S矿物已经生成较为完全。烧成温度进一步上升后熟料表面又变得较为光滑,能够观察到短柱状的C3S晶体,C3S矿物整体尺寸大,表面和缝隙存在熔剂矿物C3A和C4AF。烧成温度越高,主要矿物的质量分数越趋近于理论值,矿物形貌的发育就越完全。但烧成温度并不是越高越好,烧成温度过高会增加烧成能耗,增加液相量,减少C3S,C2S等矿物缺陷少,降低熟料与水反应时的反应活性[12]。

图6 不同烧成温度下的熟料形貌Fig.6 Clinker morphology at different temperatures
3.2 烧成时间对矿物形成的影响
煅烧时间保持不变(1 200 ℃停留10 min),在不同烧成温度分别停留10,30,50 min。不同烧成温度下,延长烧成时间的作用是不相同的。C3S和f-CaO质量分数随烧成温度和时间的变化如图7所示。1 350 ℃以上时,烧成时间相比于烧成温度对C3S的形成影响更大。在烧成时间为50 min时,烧成温度为1 350,1 400和1 450 ℃时的C3S质量分数较为接近,f-CaO质量分数略有下降;在烧成时间为10和30 min时,烧成温度从1 400 ℃上升至1 450 ℃时的C3S增加量较少;在1 350~1 450 ℃内,随着烧成时间增加,C3S质量分数增加,f-CaO质量分数下降。图7中C3S生成反应在1 300 ℃时是化学反应控制的,1 350 ℃时为过渡控制,1 400~1 450 ℃为扩散控制,实际回转窑中反应时为扩散控制[13]。考虑到试验时生料为堆积状态,增加搅拌会提高C3S的生成量和f-CaO的消耗量。因此,在1 350 ℃或1 400 ℃下是可以烧成熟料的。

图7 C3S和f-CaO质量分数与不同烧成条件的关系Fig.7 Relationship between Ca3SiO5 and f-CaOcontent and different calcination conditions
3.3 预合成硅酸二钙对硅酸三钙的影响
3.1节中证实烧成过程中发生的主要反应是C2S转变为C3S,而C3S生成速度的快慢和C3S质量分数的高低决定了熟料质量的高低,因此对C3S的生成过程进行了研究。C3S可以由CaO与SiO2反应生成或C2S与CaO反应生成。通过试剂直接配置CaO∶SiO2摩尔比为3∶1以及C2S∶CaO摩尔比为1∶1在不同烧成温度下停留30 min烧成。试验中采用的CaO与SiO2均为国药试剂,C2S为试剂预先合成的。烧成结果如图8所示。在1 350 ℃时CaO与SiO2很难直接反应生成C3S,即使将温度提升至1 450 ℃也只有少量C3S生成;CaO与C2S直接反应时的C3S生成量比CaO与SiO2直接反应的C3S生成量高;1 350 ℃时C3S质量分数在20%左右,1 450 ℃时C3S质量分数达到40%。
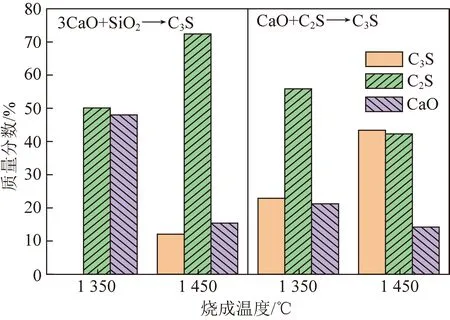
图8 不同合成路径矿物生成情况Fig.8 Mineral formation in different synthetic pathways
纯固相反应条件下C3S的生成反应是扩散控制的,其生成速率与反应温度正相关,反应温度越高则反应速率越快。C3S可以由固-固反应[14]、固-液反应和液-液反应[15]生成。图8证实了Ca2+的在固相中的扩散速率慢,f-CaO消耗速率低。因此由纯固相合成C3S温度一般较高,在1 500 ℃以上[16]。在C2S作为反应物存在时,C2S与CaO接触合成C3S的速率非常快,1 350 ℃时30 min内就能反应生成20%的C3S。预合成C2S在纯固相条件下促进了C3S生成,但C3S生成仍受制于固相反应Ca2+的扩散。
实际烧成过程中,C3S的生成往往有液相参与,液相能够显著提升Ca2+的扩散速率,因此研究了液相存在下的C3S生成。试验中的液相采用试剂合成,液相的组成为54.8% CaO+22.7% Al2O3+6% SiO2+16.5% Fe2O3,这种组成正好处于C3S-C2S-C3A -C4AF四元组分相图的最低共熔点1 338 ℃。试验过程和图8试验相似,不同之处在于增加了占反应物总质量20%的液相,结果如图9,10所示。在液相的参与下,CaO与SiO2在1 350 ℃直接反应生成C3S,10 min时C3S质量分数达到7.2%,30 min时C3S质量分数达到24%;烧成温度上升至1 450 ℃煅烧30 min时,C3S质量分数达到48.7%;在液相的参与下CaO与C2S直接反应时,在1 350 ℃,10 min时C3S质量分数达到23.2%,30 min时C3S质量分数达到30.5%,温度上升至1 450 ℃煅烧30 min时C3S质量分数达到54%。

图9 20%液相下CaO和SiO2反应产物Fig.9 Products of CaO and SiO2 reaction with 20% liquid phase

图10 20%液相下预合成CaO与C2S反应产物Fig.10 Products of pre-synthesized C2S and CaOreaction with 20% liquid phase

3.4 液相质量分数对硅酸三钙生成的影响
液相质量分数与烧成温度有关,根据铝氧率(是否≥1.38)和烧成时的温度可以计算出液相占整体反应物的比例[19]。液相量还受原料成分的影响,原料中的碱金属、碱土金属等也会增加液相的质量分数。为了保证液相质量分数准确,通过改变液相掺入量(预制液相)的形式进行试验。1 450 ℃下观察矿物生成量随液相质量分数的关系,结果如图11所示。预合成C2S转变为C3S时,随着液相质量分数的增加,C3S生成量先增加后减少,C2S生成量先减少后增加;C3A和C4AF生成量随着液相量的增加而增加,f-CaO质量分数则是随着液相质量分数的增加而减少,在液相占比为40%时f-CaO完全消失;增加液相占比导致液相量高,使得更多物料被浸润,反应更加充分,未参加反应而剩余的CaO质量分数降低,液相占比从30%提升至40%时C3S生成量降低,可见液相占比对于C3S的形成并非越高越好。即使不考虑液相过多造成熟料易结块等负面影响,液相过多也会阻碍C3S形成。C3S在液相中形成的控制因素可划分为溶解速率控制学派(苏联)、扩散速率控制学派(丹麦)和溶解+扩散控制学派(日本)[20],当液相质量分数过高时,Ca2+溶解进入液相中的速率不变,Ca2+浓度低,难以达到过饱和与反扩散,C3S结晶数量下降。当液相不存在时,溶解速率为0,C3S同样受到限制,故溶解+扩散较为符合C3S形成机制。水泥回转窑中液相质量分数通常控制在22%~26%,液相质量分数在预煅烧-烧成工艺过程中可以控制在20%~30%。

图11 不同液相质量分数下预合成C2S与CaO反应矿物形成情况Fig.11 Products of presynthesized C2S and CaO withdifferent liquid content
4 理论工艺能耗计算
通过计算预分解窑工艺和预煅烧-烧成工艺的理论熟料形成热,比较两者的差别。分析时为了方便与已有文献进行对比,采用的煤粉为(低位热值为23 614 kJ/kg,灰分为28.36%,水分为0.64%)和生料(成分)进行计算[21]。计算过程参考JC/T 730—2007 附录B中的熟料形成热理论计算方法,物料的具体参数见表2。
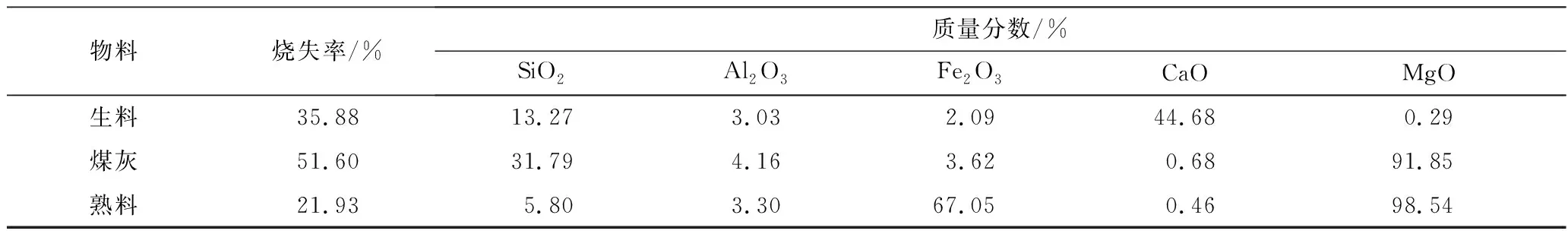
表2 生料、煤灰和熟料化学成分
由表2中数据按照率值计算公式,可以得熟料石灰饱和系数为0.92,硅酸率为2.41,铝氧率为1.76。由此可以计算出熟料主要成分质量分数:C3S为62.58%,C2S为15.65%,C3A为9.77%,C4AF为12.65%。计算中假定熟料热耗为3 188 kJ/kg,预分解窑工艺中生料入窑温度设定为900 ℃,煅烧温度为1 450 ℃,预煅烧-烧成工艺的温度分别设定为1 200 ℃和1 350 ℃。理论熟料形成热见表3(理论计算)。
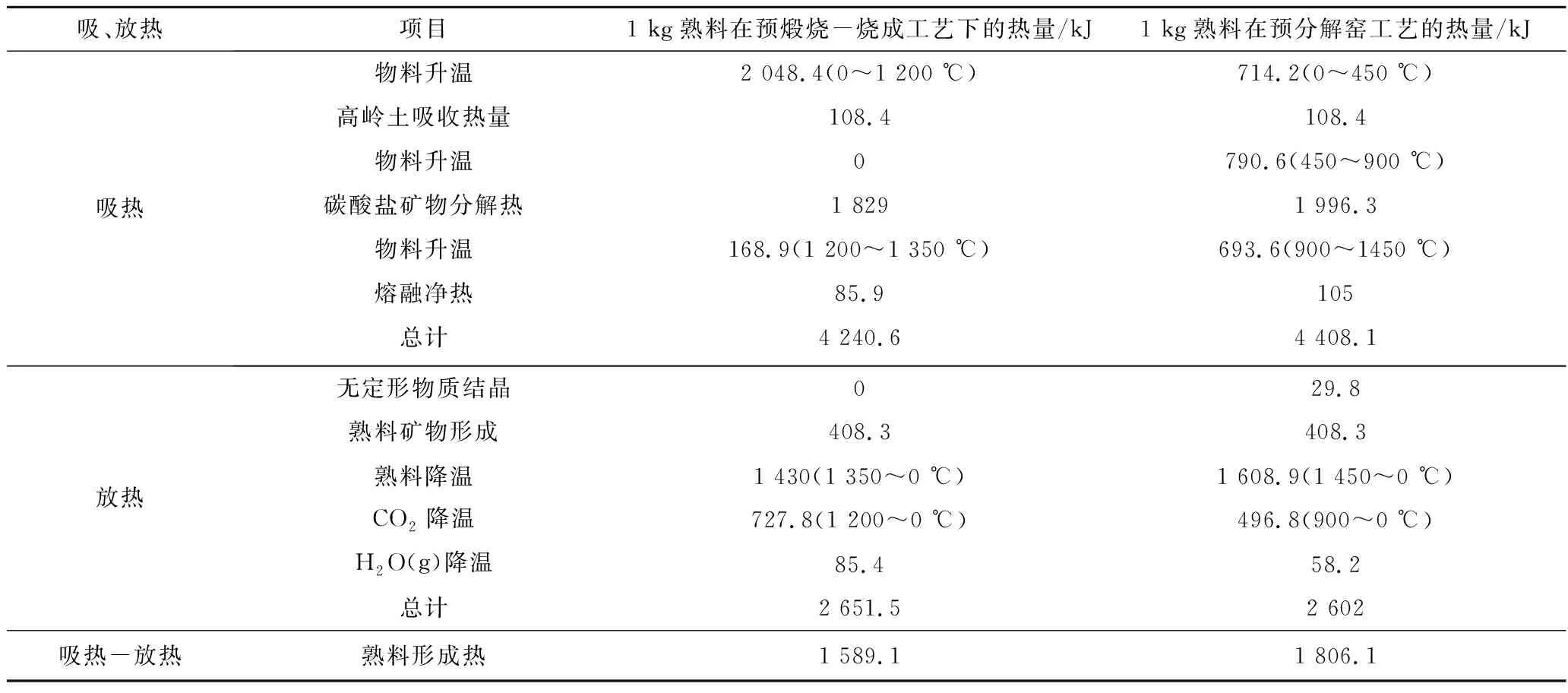
表3 预煅烧-烧成工艺与预分解窑工艺理论熟料形成热
通过对比生产1 kg的熟料所需要的热量发现,预煅烧-烧成工艺要比预分解窑工艺节省217 kJ/kg。因此,可以从理论上判定,预煅烧-烧成工艺是更加节能的,节约的能量主要来自升高温度后CaCO3分解降低的分解热耗。进一步估算预煅烧-烧成工艺排放(理论计算),选取1 000 t/d规模的2种设备进行比较,结果见表4,预煅烧-烧成装置每年能够少排放23 152 t CO2。1 000 t/d规模下川崎重工的FAKs系统相较于回转窑则是减少排放10 000 t CO2,可见流态化预煅烧-烧成工艺减排潜力巨大[22]。

表4 分段煅烧工艺与预分解窑工艺CO2排放计算
5 结 论
(1)预煅烧生成的硅酸二钙有利于硅酸三钙的形成,预煅烧-烧成工艺液相量维持在20%~30%是合适的。
(2)煅烧温度为1 200 ℃,煅烧时间维持在8~12 min,烧成温度在1 350 ℃,烧成时间30 min,可选作熟料烧成的条件。
(3)预煅烧-烧成工艺的理论熟料形成热低,1 000 t/d的装置每年能够减少23 152 t CO2,具有巨大的减排潜力。
猜你喜欢
水泥工程(2022年2期)2022-08-22
建材发展导向(2022年12期)2022-08-19
建材发展导向(2022年1期)2022-03-08
一重技术(2021年5期)2022-01-18
水泥技术(2021年6期)2021-12-31
建材发展导向(2021年20期)2021-11-20
环境污染与防治(2021年5期)2021-06-01
建材发展导向(2021年24期)2021-02-12
水泥工程(2020年2期)2020-01-04
重型机械(2019年3期)2019-08-27
