
动力学强化水合储氢技术研究进展
2023-01-06 02:43陈思远王燕鸿郎雪梅樊栓狮
储能科学与技术 2022年12期
陈思远,王燕鸿,2,3,郎雪梅,2,3,樊栓狮,2,3
(1华南理工大学化学与化工学院;2广东省燃料电池技术重点实验室,广东 广州 510640;3广东省先进绝缘涂料工程技术研究中心,广东 珠海 519175)
在资源环境问题突出和“碳达峰、碳中和”的时代背景下,寻求发展高效、清洁可持续的能源成为我国能源发展领域的重要议题。氢能是典型的绿色能源,具有燃烧热值高的特点,是化石能源3~5倍[1],成为了可替代三大化石能源的经济、有效的新能源之一。
氢能储存技术作为氢能从生产到利用的中间桥梁,是氢能发展不可或缺的一部分。氢气储存主要分为物理储存和化学储存,如图1所示,常见的方法有低温液化储氢、高压气态储氢、吸附储氢、金属合金储氢、水合物储氢等[2-8]。低温液化需要在常压下将氢气降温到20 K,液化1 kg 氢气需要消耗15.2 kWh 的电能[2],相当于储存氢气能量的30%。同时储运过程中对于容器的绝热性能要求极高,成本较高。高压储氢则是在常温下将气态的氢压缩至高压状态储存在气罐中,应用比较广泛,成本低。吸附储氢是利用材料与氢气分子间的相互作用将氢气吸附到固体物质载体中,这在一定程度上限制了储存容量;金属合金储氢条件较为温和,但需要高温进行释氢,因此需要额外的能量供给。

图1 储氢技术示意图Fig.1 Schematic diagram showing the hydrogen storage technology
笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质。常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9-10]。基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11]、二氧化碳捕获[12-15]、海水淡化[16]、气体分离[17-18]、储氢[19-21]等技术。水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21 世纪氢能储存最具发展潜力的技术[22-23]。
早期,研究人员认为氢气分子尺寸太小,不能形成水合物。直到1993年,Vos等[24]首次发现在压力0.75 GPa和3.1 GPa,温度为295 K的情况下可以形成氢气水合物,推翻了氢气分子不能稳定存在于水合物笼中的猜测。随后Dyadin[25]、Mao等[26]分别通过一系列实验和技术证实了氢气水合物的结构,并基于此提出了水合物储氢的概念;随后Mao等[27]在240~249 K、200~300 MPa条件下得到了5.3%(质量分数,下同)的储氢量的氢气水合物,满足DOE 对于车载氢燃料储氢密度的要求[28],从此水合物才作为储氢材料进入人们的视野。
然而氢气水合物生成条件过于苛刻,几乎不具有工业化应用的可能性。为此研究人员通过大量的研究发现向氢气水合物体系内引入某些添加剂可以大幅降低氢气水合物的相平衡条件,这类添加剂被称为热力学添加剂,常见的热力学添加剂有四氢呋喃[29]、环戊烷[30]、呋喃[31]、四丁基氟化铵[32]等。添加剂通过占据水合物笼的空腔稳定水合物结构,从而满足降低相平衡条件的要求。利用添加剂可将氢气水合物的相平衡从200 MPa、270 K 降到10 MPa、280.5~301.5 K[33-35]。添加剂的应用使水合物储氢具有工业化前景。
1 氢气水合物的动力学研究
添加剂的使用使水合物储氢具有了热力学可行性,然而,气体水合物的生成过程是一个多相复杂体系内气体分子和水分子相互作用的动力学过程,是其流体相向固体相转变的过程,更像是一个结晶动力学过程,包括成核和生长两个动力学过程[9]。
1.1 成核动力学
水合物成核过程是指水分子与客体分子形成超过临界尺寸的稳定水合物晶核的过程,当溶液处于过冷状态或者过饱和状态时,就有可能发生成核现象。一般成核分为两种方式。
①瞬时成核:成核在瞬间完成,此后水合物生长过程中晶核数目稳定,不再有新的晶核产生。瞬时成核对应的状态为溶液中每个微晶都是成核活性位点,当溶液处于过饱和后,都会生成水合物核,因此含有固定数量的水合物晶核。
②渐进成核:水合物边成核边生长,生长过程中伴随着成核,即水合物生长过程中晶粒数目是逐步增多的。当发生渐进成核时,成核速率与时间无关,并且符合公式(1),渐进成核速率[36]由式(1)给出。

式(1)中,A为动力学参数,c为形状参数,νh为水合物构造单元体积;σef为有效比表面能,∆μ为成核推动力,k为Boltzmann常数。
从相态来说水合物成核也可以分为均质成核和非均质成核。均质成核是指纯液体在没有其他任何杂质情况下的成核过程;非均质成核是指在溶液中存在其他物质的情况,这种成核过程在现实中更加普遍。目前有多种水合物成核模型来解释非均质水合物成核的微观机理,主要有成簇成核模型[37]、界面成核模型[38]、团簇成核模型[39]等,他们普遍认为水合物成核一般发生在气液界面,界面处所需的成核吉布斯自由能较小,并且由于界面处的主体分子浓度高,气液混合引起界面的晶体结构向内部扩散,从而导致大量晶核的出现。
出现第一个临界晶核尺寸之上的水合物簇所需时间为诱导时间。诱导时间是水合物成核的重要参数,它不是系统的基本物性参数,但诱导时间包含着关于成核的重要信息。水合物成核随机性具体表现在成核诱导时间的随机性上,诱导时间的长短和随机性主要取决于体系的驱动力(如溶液的过冷度、过饱和、成分、搅拌等),关于渐进成核的诱导期由式(2)表示。

式(2)中αd为形成的新相可探测比率,Vs为溶液初始体积,b为形状参数,Nc为生长晶体数,m为级数,G为生长常数。从式(2)可知渐进成核中的生长常数和成核常数都不能独立地控制诱导期,这表明在氢气水合物生长结晶中,微晶成核和生长过程是相伴发生的。
图2为甲烷水合物和氢气水合物生成过程中气相压力变化示意图。如图2所示,甲烷水合物是瞬时成核过程,甲烷水合物成核有一定的诱导时间,当溶液中的微晶达到临界半径后瞬时生成大量晶核并快速完成生长过程;而氢气水合物的成核方式是典型的渐进成核,无明显成核诱导时间,由式(1)可知反应初期驱动力大,渐进成核速率快,后期随着水合物层的形成,驱动力和有效比表面能降低,成核速率变慢导致氢气水合物生成速率降低。

图2 水合过程中甲烷和氢气的气相压力变化示意图Fig.2 Schematic diagram of gas-phase pressure changes in hydrophobic processes
目前的水合物成核机理模型都需要对水合物成核过程做各种假设,成核机理仍未完善,但目前广为认可的是水合物成核速率与成核推动力成正比,影响成核的变量还包括客体尺寸,表面面积、溶液状态以及搅拌程度。
1.2 生长动力学
水合物晶核形成后,体系将自发地向吉布斯自由能减小的方向发展,进入生长阶段。通常来说,水合物的生长受到传热与传质两方面的影响。
水合物的生成需要低温环境,而水合物的生成又是个放热反应,大量生成热的释放会抑制水合物的成核以及生长,甚至造成水合物的分解。对于sII型二元氢气水合物,其分解焓大小与相平衡条件、添加剂分子以及水合物中水分子间的氢键强弱等因素相关。表1给出了不同体系下二元水合物的分解焓计算结果。从表1可以看出,水合储氢过程中的水合物热不能忽略。氢气水合物的渐进式成核生长虽不会大幅提高体系温度,造成热抑制[40]。但若这些热不能及时排除,仍会使体系温度提高从而削弱水合反应速率。
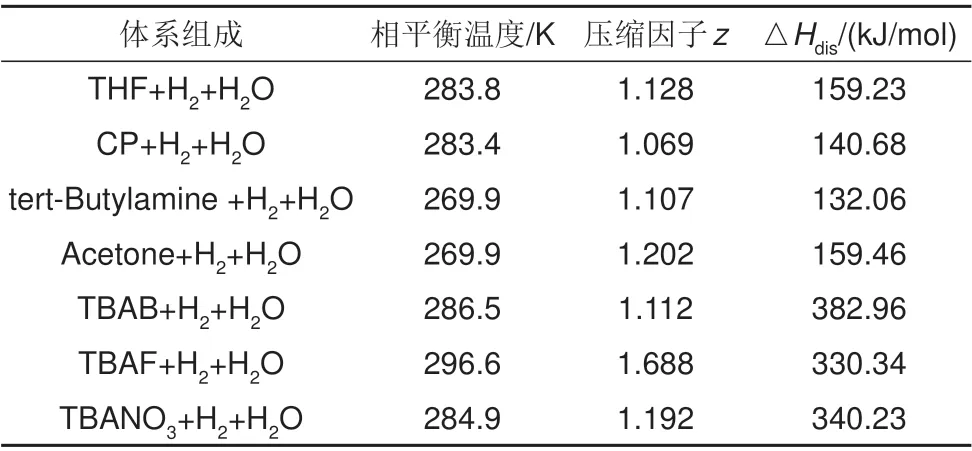
表1 II型二元氢气水合物分解焓[41]Table 1 Decompound enthalpy of sII binary hydrogen hydrate[41]
在传质方面,最初认为由于氢气分子尺寸原因,氢气水合物的生长不存在传质阻碍[42]。如表2所示,氢气分子的分子直径仅为2.3 Å(1 Å=0.1 nm),远小于甲烷、二氧化碳等气体分子直径和水合物笼直径(氢气分子与水合物孔穴直径比仅有0.3~0.5),并且氢气分子逸度很大,可以自由进出水合物笼,这也是氢气水合物难以稳定的原因之一[8]。中国石油大学(北京)陈光进教授团队等[43]用拉曼光谱研究H2、H2+CH4和H2+CO2在THF水合物生长和迁移规律,结果表明H2分子可以在5 mm厚的THF水合物层内进行渗透、迁移和储存,其中H2分子在THF水合物中渗透迁移所需要的最小分压为2.9 MPa,其扩散系数为6.1×10-12m2/s, 远大于CH4(9.4×10-17m2/s)[44]和CO2(约10-18m2/s)[45]的扩散系数,表明了水合物层对CH4和CO2具有明显传质阻力;Guillermo等[84]使用从头算密度泛函理论研究了水合物中CH4、CO2和H2分子的吸附和扩散,扩散活化能分别为1.0、0.4 和0.2 eV,种种研究表明H2分子只是比CH4、CO2等气体分子更容易在水合物层扩散,但是仍需要一定驱动力,水合储氢过程仍需考虑传质问题。
对比近年来水合储氢相关实验结果(表3),可以发现,当水合物样品量极小时,储氢密度可以达到3%以上,而当样品量超过5 g 时,储氢量就只有0.5%左右。随样品量的增加,达到100 g 以上时[32,46-47],储量仅剩0.1%。这些结果表明即使氢气分子进出水合物笼相较甲烷等大分子气体容易,但其储氢速率和储氢密度仍受气液接触面积以及氢气分子在水合物相中扩散的影响。

表3 不同样品量对水合储氢能力的影响Table 3 The effect of different sample quantities on hydrogen storage capacity
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素。增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58]。常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率。
氢气分子扩散是气相向水合物相和水合物相向气相的双向扩散,氢气分子在笼型水合物的扩散是影响水合物储氢密度的关键。分子的扩散受扩散物质的分子大小、驱动力以及扩散通道影响,通常用扩散系数来表示分子的扩散能力[59]。
驱动力是影响氢气分子扩散的关键因素,图3是在277.15 K不同驱动力下水合储氢实验数据图。如图3 所示,提高驱动力,水合储氢的初始反应速率(R1)大幅提升,从22.84 v/v·h-1提升到71.93 v/v·h-1,生成时间随之减少,同时储氢密度提高。当驱动力从19.9 MPa 增加到66.2 MPa 时,储氢量从0.345%增加到0.835%[29],小笼占据率达到了79%。可以看出驱动力是强化氢气分子扩散,促进氢气水合物成核和生长的关键因素。
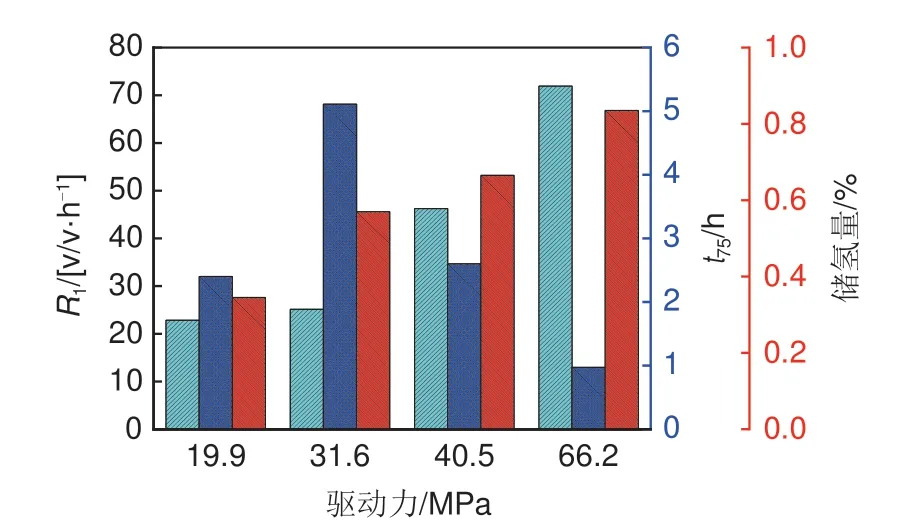
图3 277.15 K不同驱动力下水合储氢实验数据图[29]Fig.3 Graph showing hydrogen hydrate storage with different driver forces under 277.15 K[29]
此外氢气的传质还受扩散通道的影响。水合物的生成是个界面反应,水合物形成后会在气液或气固界面形成水合物膜,氢气分子必须穿透表面的水合物膜才能实现进一步水合反应;随后氢气分子在水合物笼中扩散,如图4 所示。Hasegawa 等[60]用分子动力学模拟研究了氢气分子在sII 型二元水合物中扩散的动力学过程,观察到当氢气分子在两个大笼之间扩散时,氢气分子主要通过六元环的中心扩散,扩散过程中笼结构无任何变形;但当氢气分子从一个大笼移动到小笼时,则无法通过笼的窗口进行扩散,必须要破坏部分水合物结构,即需要一个五元环断裂为氢气提供扩散通道;Gorman 等[61]也发现双氢气分子占据体系,在200~250 K 时,氢气扩散主要是通过小笼五元环氢键的断裂和重组进行扩散;王燕鸿等[62-63]采用分子动力学模拟研究了sII 型和sH 型氢气二元水合物的氢气扩散行为,他们发现水合储氢过程中边界层上的512笼将阻止氢气分子扩散到水合物相。以上研究证明氢气分子主要是通过大笼的六元环在水合物体相内进行扩散,而很难通过五元环扩散,氢气分子穿过五元环需要更大的能量来进行氢键的断裂和重组。

图4 氢气水合物扩散通道示意图Fig.4 Schematic diagram of hydrogen the hydrate diffusion channels
基于以上机理研究以及实验分析,要提高水合储氢速率和储氢密度,需要从三个方面进行强化:①提高水合储氢过程中的驱动力,如提高压力或者降低水合物形成条件等;②增大气液接触面积来强化表面扩散;③改善传质通道,强化氢气分子在传质通道的扩散。
2 氢气水合物生成动力学强化技术
2.1 提高驱动力
如前所述,提高驱动力是促进水合物成核与生长动力学的有效手段,可以通过升高压力、降低温度和添加热力学添加剂来实现。纯氢气水合物严苛的生成条件使热力学添加剂成为水合储氢过程不可或缺的部分,热力学添加剂可以参与氢气水合物的形成,改变水合物生成类型,因此使用热力学添加剂来提高水合物的储氢性能,成为众多研究者的首选。
图5为已报道的sII型[33,64]、sH型[65-66]和半笼型促进剂下[35,67]氢气二元水合物的相平衡条件。利用不同促进剂可以形成不同结构的二元氢气水合物,对于驱动力的强化作用也不尽相同。通常来说是半笼型促进剂>sII 型促进剂>sH 型促进剂。表4 总结了不同晶型二元水合物储氢理论储氢密度,半笼型添加剂对二元氢气水合物的热力学促进作用最强,有利于氢气水合物成核和生长,但是半笼型结构中,可用于储存氢气的512笼数量少,因此降低了储氢密度。综合比较分析,sII型水合物结构更适合于氢气的储存。

表4 不同晶型二元水合物储氢理论储氢密度Table 4 Theoretical hydrogen storage density of binary hydrate with different crystal forms
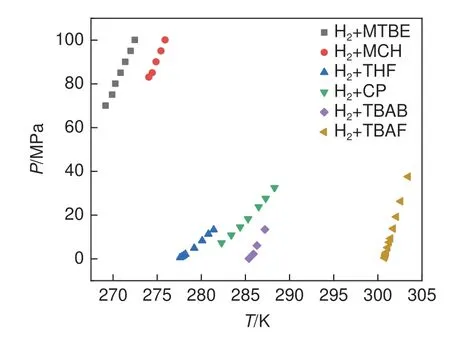
图5 不同促进剂下氢气二元水合物的相平衡条件曲线图[33,35,64-67]Fig.5 Phase diagram showing different promoters of the binary hydrogen hydrate[33,35,64-67]
除使用热力学促进剂之外,改变温度压力也是提高水合储氢性能的有效方法。表5为不同压力下水合储氢动力学参数汇总。Trueba等[31]以5.0%(摩尔分数)四氢噻吩(THT)和呋喃为促进剂,研究了二元氢气水合物与压力的关系,由表5可知,当温度稳定在277.15 K 时,压力驱动力△P从12.0 MPa增加到28.7 MPa 时,水合物生长阶段前1 小时的气体吸收速率大幅提升,初始动力学速率提高了93.8%~170.0%;而t75随压力变化较少,储氢密度相对提高,证明提高压力可以在初始阶段维持了相对较高的生长速率,压力是促进氢气水合物成核和生长的关键因素。Ogata 等[29]研究了5.6%(摩尔分数)THF 在11.4~66.4 MPa 下水合物的储氢性能,结果表明随着压力的提高,储氢速率和储氢量明显提高,其中压力主要影响反应初期前1 h内的储氢速率,后期总体反应时间相差不大,并且随着压力的增加,储氢密度增长趋势减慢,呈现饱和状态。Sugahara等[49]通过拉曼光谱、X射线衍射分析以及分解测试研究了氢气分子在THF水合物中的占据情况,结果发现即使压力提升至74 MPa,水合物储氢密度也不会显著增长,始终维持在0.7%~1%这说明提高压力增大驱动力可以提高水合储氢性能,但达到一定压力后会出现边界效应,即水合储氢性能不会出现明显改善;Burnham等[68]的分子动力学模拟认为sII 氢气水合物的大笼占据率很少会超过两个,在100 MPa 下小笼512的平均占据为0.85~1更符合现实,这也解释了边界效应的原因。
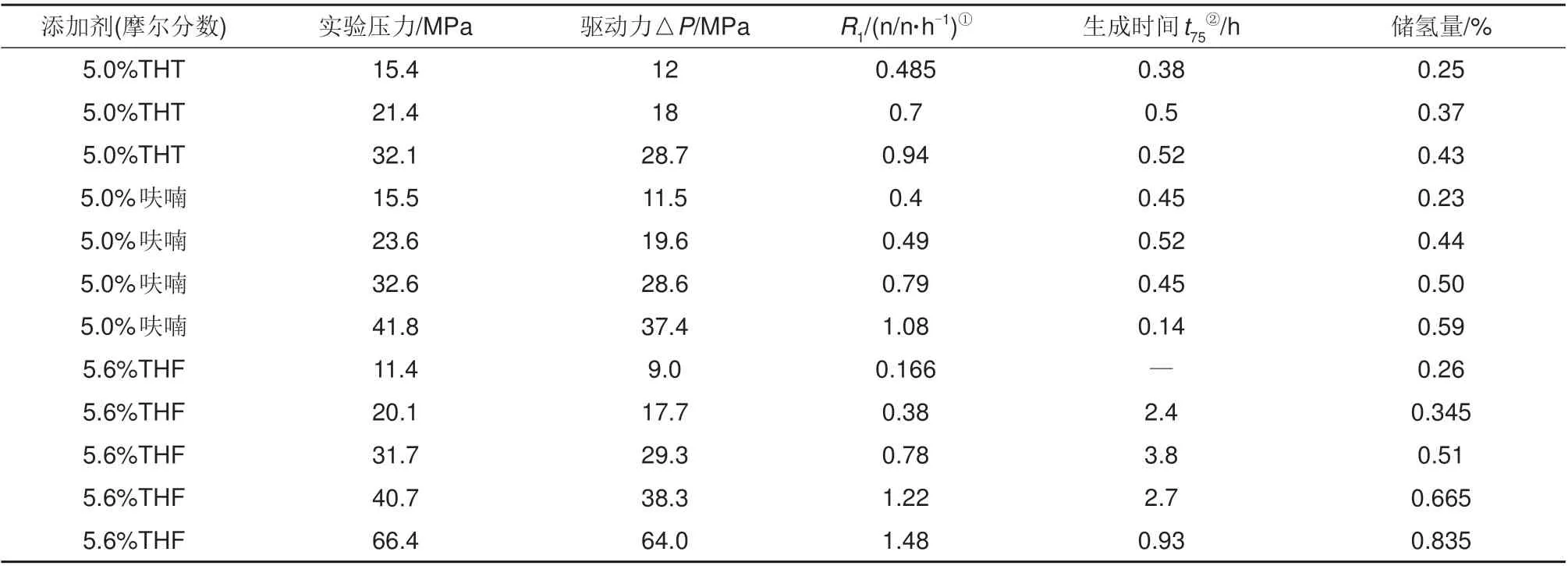
表5 在275.15 K下不同压力驱动力下水合储氢实验数据汇总表[29,31,49]Table 5 Summary table of hydrogen hydrate storage with different driving forces under 277.15 K
温度同样对水合储氢性能有明显影响,并且与压力存在显著差异。温度不仅仅影响驱动力,更与分子扩散相关。邓灿[69]研究了不同温度下氢气-四氢呋喃和氢气-环戊烷体系水合储氢,结果表明储氢量和储氢速率与温度正相关,随着温度的增高而增加,并就此建立了氢气水合物扩散模型,拟合了吸附系数以及有效扩散系数。结果表明温度对吸附系数以及有效扩散系数影响明显,高温有利于氢气分子扩散;Trinh等[70]通过分子动力学模拟也发现笼间氢气客体分子扩散的势垒随温度的升高而减小,温度高时氢气分子的扩散系数更大。Wang 等[62]的分子模拟结果也证明了,温度高时氢气分子具有更高的动能,更容易穿过水合物笼进入体相。但氢气分子的动能较高时,氢气分子也很容易从笼中逃逸;当氢气分子的动能较低时,则更容易稳定在水合物笼中,从而提高水合储氢能力。综合而言,降低温度有利于增大驱动力,但会相对降低分子扩散,因此两者之间会存在一个平衡,如何针对不同热力学促进剂体系,寻求驱动力和分子扩散的双向促进机理仍需进一步研究。
2.2 增大气液接触面积
改善气液界面通常是通过增大气液接触面积实现的,常用的方法有添加表面活性剂或机械手段、冰粉强化技术等,增强了气液两相间的传质[71]。
表面活性剂是指能使溶液表面张力显著下降的物质,具有固定的亲水亲油基团,一般可分为离子型表面活性剂、非离子型表面活性剂、两性表面活性剂等。十二烷基硫酸钠(SDS)是一种典型的阴离子表面活性剂,被广泛应用于水合物领域。Linga等[47]以5%摩尔分数THF 为热力学促进剂,首次研究了添加不同浓度SDS的氢气水合物生成动力学。实验结果表明SDS对于提高H2/THF二元水合物生成动力学影响甚微,水合物生成诱导时间没有出现明显降低。这与SDS 促进甲烷水合物生成差异甚大;之后Veluswamy 等[72]以5.6%摩尔分数THF 为热力学添加剂,深入研究了0.01%~1%质量分数的阳离子表面活性剂十二烷基三甲基氯化铵(DTAC)和非离子表面活性剂吐温-20(聚山梨酯20)存在下H2/THF 水合物生成动力学,研究结果表明0.5%的DTAC 和0.1%的吐温-20 使水合物形成速率增加约20%;随后Veluswamy 等[73]在搅拌釜反应器中研究了SDS对氢气/丙烷水合物动力学影响。SDS 浓度在5~1000 ppm(1 ppm=0.0001%)范围内,结果表明SDS 显著提高了混合水合物的形成速率,缩短了水合物的形成时间。在浓度大于100 ppm 的SDS 体系下,水合90%所需的时间从334.2 min 减少到25.5 min。表面活性剂对氢气水合物的作用取决于客体分子和所研究的系统。对于液相促进剂,表面活性剂必须要通过亲水亲油基团形成胶束,才能达到降低表面张力,扩大气液接触面积的目的;而对于气相促进剂,SDS只需要与水形成胶束,作用机理与甲烷水合物类似,因此强化效果更加明显。
表面活性剂可以在气液两相界面吸附形成胶束降低水的表面张力,也可以吸附在液体界面间形成反胶束来降低油水界面张力。Di等[30]以200 mL异辛烷为分散介质,溶解适量2-乙基己基琥珀酸酯磺酸钠(AOT)形成0.1 mol/L 的反胶束体系,如图6 所示。在以四氢噻吩为促进剂的体系下,与原本数十个小时的生成过程相比,水合物生成动力学速率明显提升,诱导时间为3 min,水合物生成时间仅为28 min,最大储氢量为0.5%。反胶束是将表面活性剂溶于非极性的有机溶剂中,当浓度超过临界胶束浓度时在有机溶剂内形成的胶束;反胶束作用机理在于表面活性剂的非极性尾向外伸入非极性有机溶剂主体中,而极性头则向内排列形成一个极性核,此极性核具有溶解促进剂分子的能力,可以有效应用到添加了热力学添加剂的氢气水合物体系。并且该研究还发现反胶束可以增强水与非水溶性分子的接触,稳定形成包裹促进剂的纳米水滴,从而增大气液接触面积,因此反胶束对于环戊烷(CP)、四氢噻吩(THT)等非水溶性促进剂的储氢效果更加明显。

图6 氢气水合物反胶束理论示意图Fig.6 Schematic diagram of hydrogen hydrate formed by reverse micelles
除使用添加剂的化学方法外,还可以通过物理方法来增强气液接触面积,使气体分子与液体接触更加充分,最终达到强化传热和传质的双重目的。
机械搅拌是水合储氢最常见的强化方式之一[31,74-75],通过叶轮旋转扩大气液接触面积,同时增加流体流动性,减少界面处成核自由能,加速水合物成核,缩短水合物形成的诱导时间。由于其设计和操作简单,机械搅拌釜在水合储氢上被广泛应用。图7对比了静态反应釜和搅拌反应器在水合储氢上的动力学性能[47,76]。结果表明机械搅拌使水合诱导时间从200 min 降至2.4 min,储氢密度也相应提高了170.97%,在低驱动力6~8 MPa 下搅拌反应器具有明显强化氢气水合物生成作用。鼓泡法是从反应釜底部产生气泡进入液相,使得气体更好地与液相接触。产生的气泡有效增大气-液两相的接触面积,并能增加气体在溶液中的溶解度。吕秋楠等[77]研究表明鼓泡法能有效增大水合物生成速率,并且生成速率随鼓泡器气体流速的增大而增大。喷雾法[78]是将水或者溶液以喷雾的形式与气体接触反应,有效增大气-液两相的传热传质效率,提高储气速率。这些动态手段都可以有效阻止水合物在界面处聚集,不断更新气液界面,对水合物的成核与生长有良好的强化效果。但随着水合物不断生成,浆液逐渐变稠,会严重影响传质效率,带来额外的能量消耗。

图7 静态和搅拌反应釜动力学性能对比图Fig.7 Comparison of kinetics performance between static and stirred tank reactors
与机械搅拌等动态方式相比,静态储氢技术是一种更加节能的方式。研究表明静态冰粉强化储氢技术可以在节约能耗的同时,大幅缩短水合物成核时间,提高水合物储氢量。冰粉储氢是将促进剂(THF、CP等)水合物研磨成粉末,再与氢气进行反应。Nagai 等[55]以四氢呋喃为热力学促进剂,研究了温度、压力和THF颗粒尺寸对于氢气水合物生成的影响,结果表明低温、高压和小尺寸的THF颗粒在动力学上更易形成氢气水合物,同时没有明显水合物生成热效应。邓灿等[56]在以环戊烷为促进剂时,对比了冰粉和溶液体系氢气水合物的形成过程。相比于搅拌溶液体系,冰粉体系的诱导时间从2.5 h降至无明显诱导时间,生成时间从30 h 降至5 h 左右,储氢量也相应提高了12.5%。该方法主要是通过物理粉碎降低水合物颗粒尺寸,从而增大氢气分子与水合物相之间的接触比表面。
与动态储氢技术相比,静态储氢技术平均储氢速率有所降低,合成时间从约450 min 增加到600 min 以上,储氢量却提高了数倍,但是静态储氢技术的低温环境以及不可循环性能一定程度上制约了其应用,因此仍需寻求更好的解决方案。
2.3 改善扩散通道
水合储氢反应初期在界面处形成水合物膜阻碍氢气分子与液相的接触,从而影响氢气水合物的进一步生成。为了改善氢气分子扩散通道,研究人员也采用了强化表面扩散、体相扩散通道等多种方式。
多孔材料具有复杂的孔隙结构和较大的比表面积,可以支撑水合物颗粒,为水合物生成提供更多的气体扩散通道,有效缩短水合物的成核及生长时间,是利用表面扩散通道强化水合储氢性能的一种有效方案。
Saha 等[79]使用多孔介质来促进氢气水合物的生成。他们以THF 为促进剂,在49、65、100、226 Å四种不同孔径的活性氧化铝、硅胶、色谱柱填料中,在270 K、6.5 MPa 的条件下,得到最大储氢量为1.0%;孔径为49 Å 的多孔介质下四氢呋喃-氢气水合物的最短形成时间仅为27 min,生成速率是四氢呋喃水合物粉末-氢气水合物的6~22倍。水合物形成时间随多孔介质孔径的增大而增长,氢气在四氢呋喃-氢气水合物中的扩散速率随介质尺寸或者水合物粒径的增大而减小。随后Su等[80]以5.56%(摩尔分数)THF 为促进剂,以高吸水性的轻度交联聚丙烯酸钠盐(PSA)颗粒为载体强化水合储氢,结果发现PSA 具有优异的储氢性能,仅60 min 即可达到90%的储氢量。基于气体扩散通道的理念,Zhao 等[81]提出将氢气分子放入一维碳纳米管中的新思路,理论预测了常温常压下低维氢气水合物的形成。研究发现,与体相水合物不同,一维氢气水合物中的氢气分子形成准一维分子链被包络在一维冰纳米管内,氢气分子链可以像液体一样自由地沿轴向流动,H2分子的轴向扩散系数要比溶液中的扩散系数大一个数量级。
以上研究是通过分散水合物相、降低传质阻力层的方式来减少表面扩散距离,强化表面扩散通道来提高氢气水合物的形成速率。这些方法虽然在一定程度上提高了水合储氢速率,但由于热力学促进剂分子始终占据水合物大笼,也相对限制了水合储氢密度的发展。华南理工大学王燕鸿等[63]提出了“沸石冰”的思路,即预先生成丙烷水合物,之后将丙烷脱除得到类沸石水合物结构;结果表明在264.3 K、10.34 MPa 下“沸石冰”结构储氢无明显诱导时间,在20 min 左右即完成了水合物生长过程,最终储氢容量为1.18%,同时拉曼光谱测试结果也验证了氢气分子进入了丙烷形成的水合物笼内;与丙烷-氢气混合气体水合物储氢相比,“沸石冰”储氢成核和生长时间更短,并且获得了更高的储氢容量。“沸石冰”储氢的目的在于将水合物笼中的大笼空出,为氢气分子的扩散提供了传质通道。但这种方法仅适合于气体分子充当热力学促进剂的的体系,对于液相有机促进剂无明显作用。Lee 等[82]在270 K、12 MPa 的压力下进行了0.2%(摩尔分数,下同)、0.7%和5.6%THF 溶液储氢实验,结果表明0.2%THF 体系水合反应速率最快,在60 min即反应完全,5.56%THF体系水合反应速率最慢,生成时间为100 min,并且储氢量仅为1.76%,远低于0.2%THF 体系所达到的3.8%储氢密度。他们将这种现象称之为“调谐效应”,即减少热力学促进剂浓度,空出部分水合物大笼,达到既可以稳定氢气水合物结构,又可以为氢气分子进入体相占据水合物大笼提供扩散通道的目的,从根本上提高了理论储氢量。Kim 等[83]随后基于此提出CGC(critical guest concentration)的概念,即临界客体浓度:达到最大储氢能力的液相促进剂浓度,低于此浓度可能无法使水合物结构稳定,CGC 是氢气水合物调谐效应的一个重要指标。Sugahara等[48]将180 μm 粉状冰与固态THF 相混合,制备0.54%~5.58%的固体混合物,在255 K、70 MPa下储氢,Raman 光谱分析显示大笼中H2的峰值强度随着THF 摩尔分数的进一步降低而增加,这种H2占据水合物大笼的行为与Lee等人[82]报道的“调谐效应”相似。但目前关于调谐效应仍存在争议,如何使调谐效应更好地应用是今后需要重点关注的方向。
图8总结了氢气水合物强化技术对应的储氢量和储氢时间。静态溶液体系低储氢量(约0.03%)最低、储氢时间(约700 min)最长。冰粉强化储氢技术则具有较高的储氢量,但储氢速率较机械搅拌慢;多孔介质等则可显著提高储氢速率但储氢量仍有不足,其中调谐效应具有最佳的储氢量(约1.8%)和较短的储氢时间(约120 min)。

图8 不同强化技术下氢气水合物储氢量和储氢时间的示意图Fig.8 Schematic diagram of hydrogen storage capacity and formation time under different enhancement technologies
3 结论与展望
利用水合物作为储氢材料具有成本低、安全可靠等优势,但储氢密度和储氢速率仍是未来需要解决的难点。传统的提高驱动力、增大气液接触面积以及改善通道等动力学强化技术对水合物储氢性能提升均有一定的作用,但不同的强化技术产生的促进效果不同。目前单一的促进手段仍不能很好地从根本上解决水合物储氢所面临的问题。基于水合物成核生长动力学理论和已有的研究成果,未来水合物储氢可从以下几方面开展研究。
(1)深化氢气水合物的成核与生长、稳定机制研究。目前对水合储氢机理方面的共识是氢气分子可以通过六元环进入水合物笼中,但很难穿透五元环,需要很大的动能才能进出五元环,同时又容易逸出而无法稳定存在于水合物笼中。针对此类问题未来需要完善水合储氢的相关机理研究,以期从理论角度找到降低氢气分子在小笼扩散的能量壁垒,同时抑制氢气分子大笼逸出的有效途径。
(2)优化反应过程。实验发现氢气水合物生成过程中,降温有利于增大驱动力促进成核,升温则有利于氢气在笼中的扩散。提高氢气水合物的储气密度,可以根据不同的反应阶段改变反应条件,达到成核和生长同时促进的目的。
(3)寻找高效促进剂。目前最为实际的提高驱动力的方法仍是添加热力学添加剂,但目前已知的添加剂如四氢呋喃、环戊烷等热力学平衡条件仍无法满足氢气水合物高效生成所需的驱动力,未来对于热力学添加剂的研究仍需集中在寻找新型高效的热力学添加剂上。在动力学促进剂中,目前尚无有效的动力学促进剂,利用胶束理论、吸附理论和毛细管理论等探寻新型动力学添加剂与方法,如各种氨基酸等,将添加了热力学促进剂的溶液均匀分散,强化水合储氢生成动力学。
(4)耦合动力学强化技术。目前扩散强化如冰粉、多孔介质等在提高水合储氢密度和速率方面均有明显优势,但受制备技术的限制,很难得到分子级别的颗粒。而沸石冰可以提高体相内的扩散通道。因此,可以考虑将二者耦合,改善水合物颗粒间以及体相内的传质。
猜你喜欢
橡胶科技(2022年10期)2022-11-03
橡胶科技(2022年5期)2022-07-20
轮胎工业(2022年2期)2022-07-19
中国特种设备安全(2022年4期)2022-07-08
中国特种设备安全(2022年4期)2022-07-08
合成材料老化与应用(2022年3期)2022-06-27
无机盐工业(2022年3期)2022-03-11
腐植酸(2020年5期)2020-12-20
World Journal of Clinical Cases(2019年4期)2019-04-16
科技创新与品牌(2019年12期)2019-02-06
