
Compound heterozygous p.L483P and p.S310G mutations in GBA1 cause type 1 adult Gaucher disease: A case report
2023-01-04 07:59XiaoLingWenYaoZiWangXiaLinZhangJiaQiangTuZhiJuanZhangXiaXiaLiuHaiYanLuGuoPingHaoXiaoHuanWangLinHuaYangRuiJuanZhang
World Journal of Clinical Cases 2022年36期
Xiao-Ling Wen, Yao-Zi Wang, Xia-Lin Zhang, Jia-Qiang Tu, Zhi-Juan Zhang, Xia-Xia Liu, Hai-Yan Lu, Guo-Ping Hao, Xiao-Huan Wang, Lin-Hua Yang, Rui-Juan Zhang
Xiao-Ling Wen, Jia-Qiang Tu, Department of Hematology, The First People’s Hospital of Yibin,Yibin 644000, Sichuan Province, China
Xiao-Ling Wen, Yao-Zi Wang, Zhi-Juan Zhang, Xia-Xia Liu, Lin-Hua Yang, Department of Hematology, The Second Hospital of Shanxi Medical University, Taiyuan 030001, Shanxi Province, China
Xia-Lin Zhang, Rui-Juan Zhang, Department of Hematology, The Third Hospital of Shanxi Medical University, The Shanxi Bethune Hospital, The Shanxi Academy of Medical Sciences,The Tongji Shanxi Hospital, The Shanxi Medical University, Taiyuan 030032, Shanxi Province, China
Hai-Yan Lu, Guo-Ping Hao, Xiao-Huan Wang, Department of Hematology, The Children’s Hospital of Shanxi, Taiyuan 030006, Shanxi Province, China
Abstract BACKGROUND Gaucher disease (GD) is caused by a GBA1 gene mutation that leads to decreased acid β-glucosidase activity [glucocerebrosidase (GCase)]. This study aimed to identify and characterise compound heterozygous mutations in GBA1 in a patient with type 1 GD.CASE SUMMARY Here, we report a rare adult-onset type 1 GD in a 46-year-old female patient with clinical manifestations of giant spleen, thrombocytopenia, and bone pain, diagnosed by enzymatic and genetic testing. Enzymology and whole exome sequencing revealed heterozygous missense mutations in exon 10 c.1448T>C (p.L483P) and exon 7 c.928A>G (p.S310G) of GBA1. The latter was first reported in patients with GD. Structural modelling showed that p.S310G and p.L483P were distant from the GCase active site. The p.S310G mutation in domain 1 may decrease stability between the α2 and α3 helices of GBA1. The p.L483P mutation in domain 2 reduced the van der Waals force of the side chain and disrupted the Cterminal β-sheet. The patient was treated with imiglucerase replacement therapy, and her condition was stable.CONCLUSION The p.L483P/p.S310G novel compound heterozygous mutation underlies type 1 GD and likely affects GCase protein function. This is the first description of p.S310G being associated with mild type 1 GD in the context of a coinherited p.L483P mutation.
Key Words: Gaucher disease; Parkinson’s disease; Lipid metabolism; Molecular mechanism; Case report
INTRODUCTION
Gaucher disease (GD) is a rare autosomal recessive hereditary lysosomal storage disease with a global incidence of approximately 0.4/100000-5.8/100000[1]. It arises fromGBA1gene mutations, resulting in the accumulation of glucosylceramide in the reticuloendothelial system, leading to anaemia, low platelet counts, and damage to the liver and spleen.GBA1(RefSeq: NG_009783.1) located at chromosome 1q21, spans 7.6 kb with 11 exons. There are 459 reportedGBA1mutations, including point mutations, splicing, insertions and deletions, frameshift mutations, and recombination alleles; however, not all are pathogenic[2]. GD is heterogeneous and is classified into three types based on neurological severity[3,4]. In type 1, neuronal features are not observed, and patients can be asymptomatic and present at any age[5]. Both type 2 and 3 GD have neurological involvement and are present in infancy, but only type 2 GD results in early death.
Surprisingly,in vitroexpression experiments have demonstrated that theseGBA1mutations are relatively stable and active[6,7].In vivo, glucocerebrosidase (GCase) is synthesised in the rough endoplasmic reticulum (ER) and then transported to the lysosome.GBA1mutations cause GCase to exit the ER and are destroyed by the ubiquitin-proteasome system[8]. Thus, GD patients have reduced amounts of acid β-glucosidase (GCase) in their lysosomes.
The current study examined the aetiology of type 1 GD in an adult female patient and found double heterozygosity for c.1448T>C (p.L483P) and c.928A>G (p.S310G) mutations inGBA1. The p.L483P mutation is common in Asian populations, and homozygous p.L483P mutations result in a severe GD phenotype with neurological manifestations and early death[9]. The patient with the heterozygous mutation p.L483P/p.S310G had a milder phenotype and did not have clinical GD symptoms until adulthood. The p.S310G mutation has only been reported in Parkinson’s disease (PD)[10]. However, its molecular pathogenesis remains unclear. In the current study, the effects of p.L483P/p.S310G on the clinical manifestations of GCase and GD were preliminarily explored through gene diagnosis and protein structure analysis.
CASE PRESENTATION
Chief complaints
A 46-year-old Chinese woman was admitted to the hospital in December 2019 because of progressive enlargement of the spleen for more than 3 years and a progressive decrease in platelet count for more than 9 mo.
History of present illness
The patient presented with asymptomatic splenomegaly (ultrasound: Spleen length, 154 mm; width, 41 mm) in December 2016. In December 2018, the patient complained of persistent right hip joint pain without mobility problems. Radiographic studies did not reveal significant hip joint or femoral abnormalities. In March 2019, a review ultrasound revealed an enlarged spleen (length, 153 mm; width, 57 mm) and thrombocytopenia (52 × 109/L). In November 2019, another ultrasound showed continuous spleen enlargement (length, 249 mm; width, approximately 66 mm) and further decreased platelet count (40 × 109/L).
History of past illness
She was hepatitis B surface antigen-positive in December 2016.
Personal and family history
Except for her mother’s PD diagnosis, her family history was insignificant.
Physical examination
Megasplenomegaly was noted 18 cm below the ribs, the liver was not palpable under the ribs, and the nervous system examination was normal.
Laboratory examinations
Complete blood counts showed white blood cells 3.59 × 109/L [normal: (4-10) × 109/L], haemoglobin 102 g/L (normal: 110-160 g/L), and platelets 34 × 109/L [normal: (100-300) × 109/L]. Bone marrow cytomorphological examination revealed that Gaucher-like cells accounted for 4.4% (Figure 1). The GCase activity of the peripheral blood leukocytes was 3.8 nmol/(mg·h) (reference range: 10-25 nmol/mg ∙ h). TheGBA1gene test showed heterozygous mutations c.1448T>C (p.L483P)/c.928A>G (p.S310G) (Figures 2 and 3). The patient was diagnosed with GD (type 1). The father was a p.L483P heterozygous carrier, and the mother was a p.S310G heterozygous carrier. The younger sister did not harbor any mutations. Predictions made by PolyPhen-2, SIFT, and MutationTaster suggested that the p.L483P and p.S310G mutations might be pathogenic. According to the American College of Medical Genetics and Genomics guidelines[11], p.L483P was evaluated as a pathogenic variant, and p.S310G was probably a pathogenic variant.
Imaging examinations
Bone mineral density testing of the thoracolumbar and bilateral hip joints revealed decreased bone mineral density; computed tomography showed bilateral hip joint changes; and magnetic resonance imaging showed an abnormal signal in the medullary cavity of the lower segment of the right femur, bone infarction, and abnormal signal in the medullary cavity of the left lower femur.
Protein structure analysis
GCase belongs to the GH30 family of glycoside hydrolases. In contrast to the traditional three-domain classification method, domains I and II were reclassified as domain 2, and domain III was classified as domain 1[12,13]. The GCase protein contains two domains, in which the active sites, E274 and E379, are located in the β-folded plates β4 and β7 of domain 1. The mutation point p.S310G was located in α-helix α4. However, it appears to be closer to E274 in the secondary structure. In the three-dimensional structure, p.S310G was located at the bottom of α4 and E274 at the top of β4. The distance between the two was relatively large. The mutation point p.L483P was located at the β-pleated-sheet βs6 in domain 2, which was far away from the two active centres. Therefore, being far away from the GCase active centres, the mutation points p.S310G and p.L483P were predicted not to have a significant impact on the catalytic properties of the protein.
The p.S310G mutation is located in α-helix α4. In the wild type, the C-β atom of the side chain of the Ser residue formed a van der Waals force with the C-γ2 atom of the Val253 side chain and the C atom of the Leu307 main chain. In addition, the N atom of the Ser residue main chain formed a hydrogen bond with a length of 3.5 Å and 3.2 Å with the O atom of the Thr306 and Leu307 main chains, respectively; the O atom of the main chain formed a hydrogen bond with a length of 3.3 Å with the N atom of the His313 main chain. After Ser310 was mutated to Gly310, the hydrogen bond of the main chain remained, while the van der Waals force formed by the side chain and surrounding residues disappeared. The p.S310G mutation weakened the force between this point and Val253, which is located above the α helix α3, therefore might affect the stability between α2 and α3 (Figure 4).
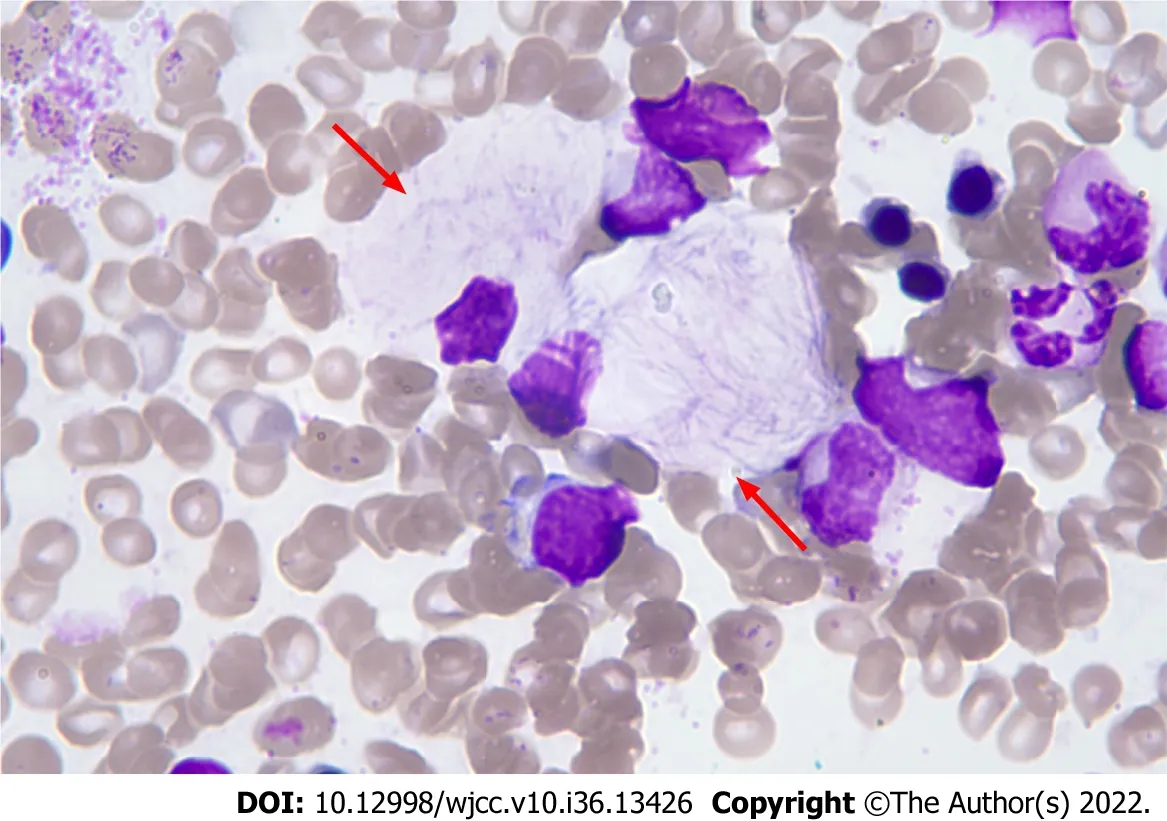
Figure 1 Bone marrow cytomorphology. Haematoxylin-eosin stain (magnification: 100 ×).
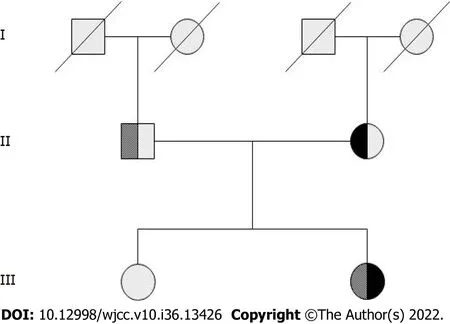
Figure 2 Family pedigree of the proband. The first-generation members have no information. The second generation is the father of proband is heterozygous for exon 10 c.1448T>C (p.L483P) mutation, and the mother is heterozygous for exon 7 c.928A>G (p.S310G) mutation. The third generation is the proband who was compound heterozygous for p.L483P/p.S310G. Her sister was normal and did not carry either mutation.
p.L483P was located in the loop region connecting βs5 and βs6 (near βs6). In the wild type, the side chain of Leu483 formed abundant van der Waals forces with the surrounding residues. The C-δ1 atom of its side chain could form van der Waals forces with the C-δ2 atom of Leu104, C-γ1 atom of Val499, Cγ1 atom of Val507, and C-δ2 atom of Leu509, and the C-δ2 atom could form van der Waals forces with C-γ atom of Asn501 and C-β atom of Ser523. Those van der Waals forces were formed because of the interactions between the side chains. When Leu483 mutated into Pro483, the N-Cα rotation of Pro was bound by the pyrrolidine ring in its structure, thereby having less conformational freedom. This structure limited the diversity of its spatial conformation, especially in the loop region. The existence of Proline could help stabilise the loop region, which was originally more flexible. Therefore, theoretically, the p.L483P mutation located in the loop region should have enhanced the stability of this region. However, owing to side chain changes, the original 6 van der Waals forces were reduced to 3, and the retained van der Waals forces were formed between C-γ of Pro483 and C-δ2 of Leu104 and C-γ1 of Val507 and one between C-β and C-γ1 of Val499. Thus, it can be seen that the introduction of Pro stabilised the conformation of the loop region, but the reduced van der Waals forces of the side chain might also affect the function of the protein structure (Figure 5).
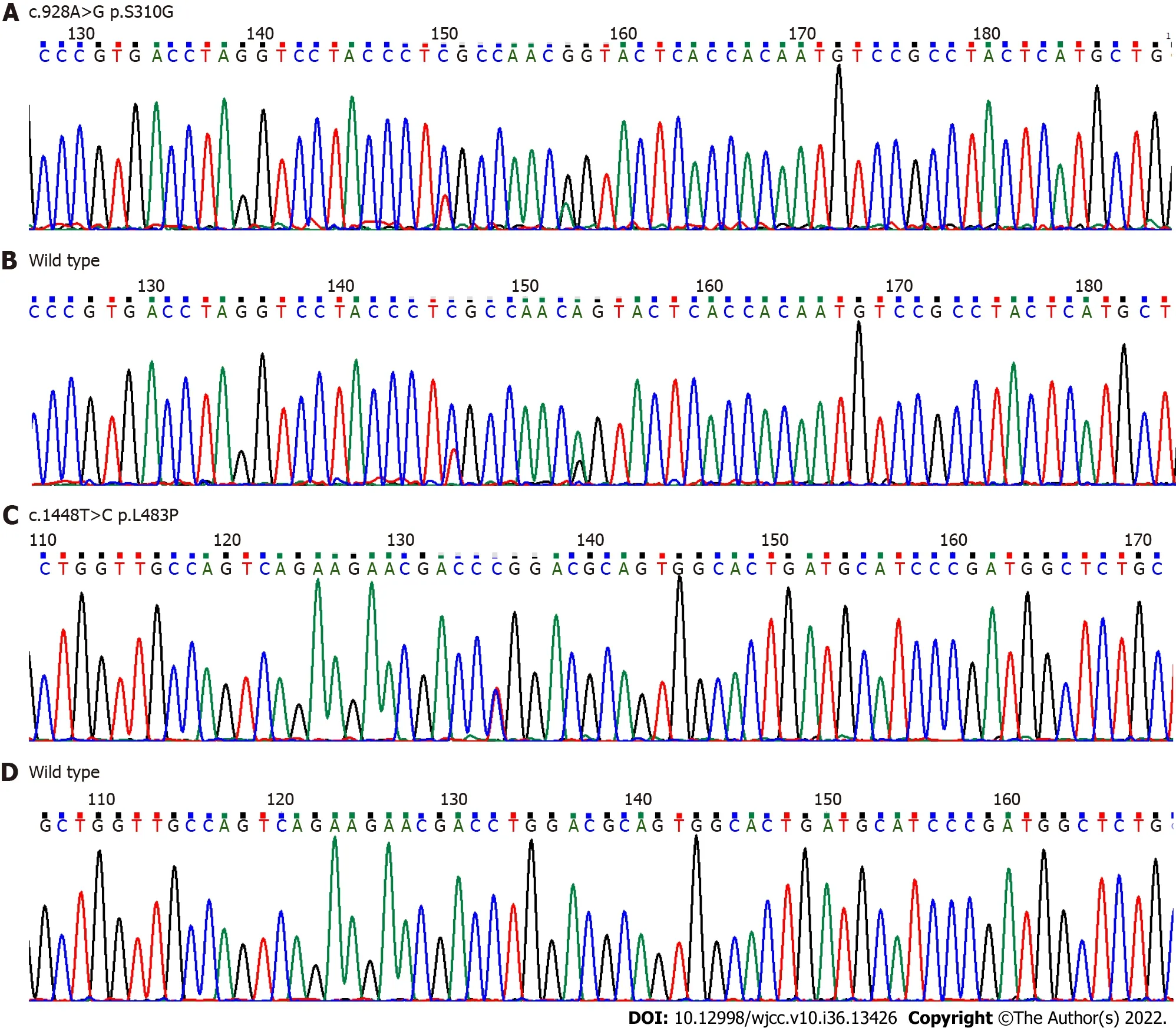
Figure 3 DNA sequencing analysis of glucocerebrosidase gene. A and B: Exon 7 c.928A>G (p.S310G) novel heterozygous missense mutation compared to the corresponding wild-type sequence; C and D: Exon 10 c.1448T>C (p.L483P) heterozygous mutation compared to the corresponding wild-type sequence.
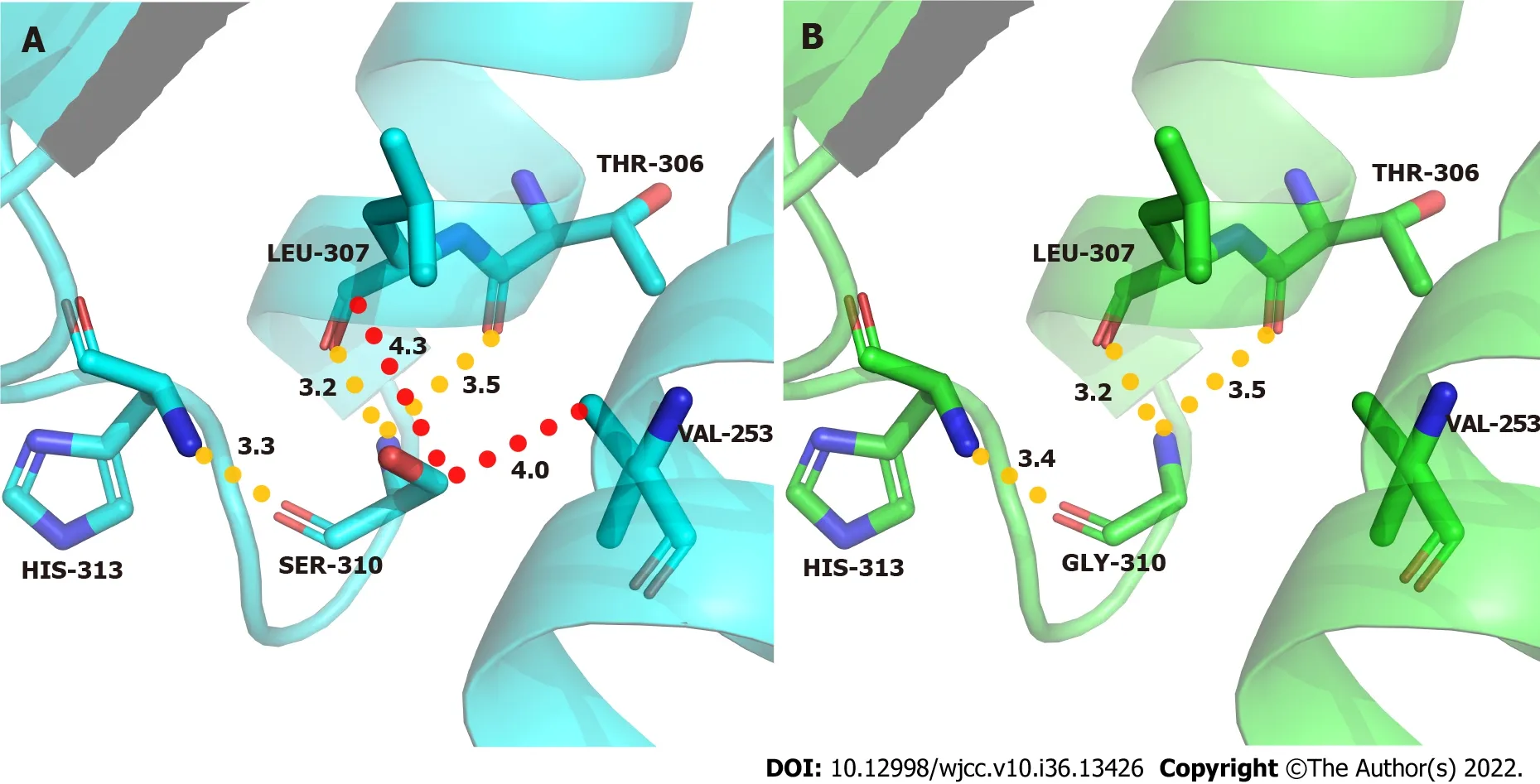
Figure 4 Molecular contacts of residue 310. A: Wild-type acid β-glucosidase protein; B: Mutant type.

Figure 5 Molecular contacts of residue 483. A: Wild-type acid β-glucosidase protein; B: Mutant type.
FINAL DIAGNOSIS
GD (type 1).
TREATMENT
In January 2020, imiglucerase was administered intravenously (45 U/Kg, once every 2 wk).
OUTCOME AND FOLLOW-UP
The bone pain was relieved, the blood routine was rechecked several times, and the platelet count was continuously lower than 30 × 109/L. No significant improvement was observed in the spleen size.
DISCUSSION
GD is a rare inborn metabolic error secondary toGBA1gene mutations that leads to reduced GCase activity. The substrate glucocerebroside (also known as glucose ceramide) accumulates in the macrophage lysosomes to form Gaucher cells. Gaucher cells accumulate widely in the liver, spleen, bones, lungs, brain, and other tissues and organs, resulting in the progressive worsening of dysfunction. Based on different clinical manifestations, GD can be divided into three types, of which type 1 (nonneuropathic) is the most common, accounting for approximately 95% of cases[4]. GD can occur at all ages with widely varying clinical manifestations. No primary central nervous system involvement is found in type 1 GD, and enzyme replacement therapy is the main treatment.
A previous study suggested thatGBA1mutations are closely associated with PD[14]. Carriers ofGBA1gene mutations, such as p.N370S and p.L483P, were found to increase the risk of PD disease by 2.16 times, with the characteristics of early-onset age and declined cognitive ability. A possible pathogenic mechanism is that these mutations increase α-synaptic nucleoprotein aggregation[15-18]. The coexistence of p.L483P with recombinant alleles, splice variants, or known severe missense alleles, such as p.D488H and p.R159W, has been reported to commonly lead to neuronopathic GD. When coexisting with mild mutations, such as p.N409S and p.P305A, it manifests as non-neuronopathic GD[9,19]. p.L483P has long been considered a serious pathogenic mutation; however, p.L483P has been reported to be associated with delayed onset of neurological symptoms in type 3 patients with Japanese patients[20], which may be related to the possible genetic heterogeneity among different ethnic groups. Enzyme replacement therapy can significantly improve haematological and other systemic symptoms in patients with type I GD, but its efficacy in patients with type 3 GD remains controversial. It has been reported in the literature that some type 3 patients developed symptoms of nervous system involvement after a median treatment period of 7.6 years[21]. In this study, the patient was treated with imiglucerase for 2 years, the platelet count continued to fail to recover, and the splenomegaly did not improve significantly. The proband with type 3 GD is the most common pathogenic mutation, p.L483P, and her mother carries the p.S310G mutation, which has been confirmed to be a patient with PD. Therefore, this proband is a high-risk patient for PD and should be aware of the combination of neurological symptoms. The p.S310G mutation has only been reported in PD, and to the best of our knowledge, this is the first report of this mutation in patients with GD. However, its molecular mechanism of action and significance in GD remain unclear. In our patient with type 1 GD, the p.L483P mutation alone was not sufficient to result in the GD phenotype, suggesting that the p.S310G mutation contributed to the manifestation of type 1 disease.
In this study, to further understand the significance of p.L483P/p.S310G compound heterozygous mutations in GD, GCase protein structure analysis was conducted to probe the pathogenic characteristics of p.L483P/p.S310G mutations at the protein level. GCase belongs to the glycoside hydrolases GH30 family. The GH30 protein structure was first defined in 2010[13]. Based on the overall characteristics of GH30 family proteins, this study divided the GCase protein into two domains.GBA1mutations can significantly influence the structure and function of the protein, including reducing the stability of domain III, premature termination of translation, and interference with catalytic activity[22-24]. Protein structure simulation revealed that p.L483P and p.S310G mutation points were far from the GCase active centre, suggesting that they should have little effect on the catalytic properties of the GCase protein. This also conformed to the patient’s late-onset age with a mild clinical phenotype. Located in domain 2, the p.L483P mutation is believed to affect the structure and function of the protein by reducing van der Waals forces of the side chain. Earlier studies have suggested that the p.L483P mutation could lead to decreased enzymatic activity[25], affect the catalytic activity of GCase[26], disrupt the hydrophobic core and domain folding[17], or alter protein stability by reducing intramolecular hydrogen bonding[22]. Our study is different from the previous study, which further enriches the understanding of the impact of the p.L483P mutation on the protein structure. In this study, we demonstrated the first observed influence of the p.S310G mutation on the GCase protein. The p.S310G mutation in domain 1 could decrease the stability between α2 and α3 of the α-helix of the GCase protein, thus affecting the function of the GCase protein.
CONCLUSION
The current study verified that the p.L483P/p.S310G novel compound heterozygous mutation was the cause of GD. This mutation caused the disease probably by interfering with the biological function of the GCase protein. A follow-up study will be performed to assess the risk of developing PD in patients with the p. L483P variant.
FOOTNOTES
Author contributions:Zhang RJ, Wen XL and Zhang XL designed the study. Zhang XL and Wen XL performed the experiments. Wen XL and Wang YZ drafted the manuscript; and all authors have contributed to the revision of the manuscript.
Supported byShanxi Key Research and Development Project, No. 201903D321133; Shanxi Bethune Hospital’s Talent Introduction Scientific Research Start-up Fund Project, No. 2021RC038 and 2021RC017.
Informed consent statement:Informed written consent was obtained from the patient for the publication of this report and any accompanying images. This study was approved by the Ethics Committee of the Third Hospital of the Shanxi Medical University (approval no. SBQKL-2021-052) and in accordance with the principles of the Declaration of Helsinki.
Conflict-of-interest statement:All the authors report no relevant conflicts of interest for this article.
CARE Checklist (2016) statement:The authors read the CARE Checklist (2016), and the manuscript was prepared and revised according to the CARE Checklist (2016).
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORCID number:Xiao-Ling Wen 0000-0001-7300-4027; Rui-Juan Zhang 0000-0001-7300-4027.
S-Editor:Wang JJ
L-Editor:A
P-Editor:Wang JJ
 World Journal of Clinical Cases2022年36期
World Journal of Clinical Cases2022年36期
- World Journal of Clinical Cases的其它文章
- Liver injury in COVID-19: Holds ferritinophagy-mediated ferroptosis accountable
- Amebic liver abscess by Entamoeba histolytica
- Living with liver disease in the era of COVID-19-the impact of the epidemic and the threat to high-risk populations
- Cortical bone trajectory screws in the treatment of lumbar degenerative disc disease in patients with osteoporosis
- Probiotics for preventing gestational diabetes in overweight or obese pregnant women: A review
- Effectiveness of microwave endometrial ablation combined with hysteroscopic transcervical resection in treating submucous uterine myomas