
Liver injury in COVID-19: Holds ferritinophagy-mediated ferroptosis accountable
2023-01-04 07:59FengJuJiaJingHan
World Journal of Clinical Cases 2022年36期
Feng-Ju Jia, Jing Han
Feng-Ju Jia, Jing Han, School of Nursing, Qingdao University, Qingdao 266071, Shandong Province, China
Abstract Even in patients without a history of liver disease, liver injury caused by coronavirus disease 2019 (COVID-19) is gradually becoming more common. However, the precise pathophysiological mechanisms behind COVID-19's liver pathogenicity are still not fully understood. We hypothesize that inflammation may become worse by cytokine storms caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Elevated ferritin levels can initiate ferritinophagy mediated by nuclear receptor coactivator 4 (NCOA4), which leads to iron elevation, and ferroptosis. In COVID-19 patients, ferroptosis can be restricted to reduce disease severity and liver damage by targeting NCOA4-mediated ferritinophagy. To confirm the role of ferritinophagy-mediated ferroptosis in SARS-CoV-2 infection, further research is required.
Key Words: COVID-19; Liver injury; Ferritinophagy; Ferroptosis; Iron; SARS-CoV-2
INTRODUCTION
The coronavirus disease 2019 (COVID-19) pandemic is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and exhibits a wide range of severity, from mild symptoms to severe presentation and death. Although respiratory symptoms are more frequent, signs of hepatic involvement have gradually come to light[1-4]. Aside from respiratory complications, the most common complication of SARS-CoV-2 infection is liver injury, which has been reported in up to 50% of cases[5-7]. Expanding prove shows a close relationship between anomalous liver function and the severity and mortality of the illness[8,9]. Derangement of alanine aminotransferase/aspartate aminotransferase levels is a key indicator of liver damage in COVID-19, accompanied by marginally elevated bilirubin levels, which are commonly used to diagnose hepatic injury such as acute hepatitis, steatosis, portal inflammation, granulomas, thrombotic bodies, and biliary pathology[10-12]. The raised liver catalysts are not only associated with severe disease and longer disease duration, but also common in the early stage of the COVID-19 pandemic[13]. However, the precise cause of liver damage is still unclear[14,15].
SARS-COV-2 INFECTION-ACTUATED LIVER INJURY IN COVID-19
The following potential mechanisms have been suggested that may be responsible for hepatic injury: SARS-CoV-2 causing direct harm to hepatocytes and biliary epithelium, indirect damage prompted by an exaggerated cytokine storm, and/or drug-incited hepatoxicity[10]. Patients with COVID-19 may experience liver dysfunction as a direct result of viral infection. A previous study showed the liver pathology of SARS patients and found SARS-associated coronavirus in liver tissues, suggesting that hepatic impairment was caused by viral infection in the liver[16]. Angiotensin converting enzyme 2 (ACE2) has recently been discovered as the SARS-functional CoV's host cell receptor, facilitating the entry of SARS-CoV-2 into cells[17-20]. The spike (S) protein of SARS-CoV-2 may also be broken down by transmembrane protease serine 2 (TMPRSS2), which makes it easier for the virus to fuse with cellular membranes[21]. Although they are expressed in many organs, including the liver, ACE2 and TMPRSS2 are expressed at varying amounts in different cell types, with cholangiocytes expressing them at a higher level than hepatocytes[22,23]. It has been demonstrated that hepatocyte and cholangiocyte organoids are receptive to SARS-CoV-2 infection[24]; however,in vivoconfirmation of this phenomenon is still pending[25]. Ultrastructural and histological analysis have shown that hepatocytes exhibit a characteristic SARS-CoV-2 infected lesion[26]. In-depth proteomic analysis of autopsy tissue recently produced new data that showed little evidence of virus replication in the liver[27]. Additionally, an autopsy of a COVID-19 patient demonstrated that the liver tissue was free of viral inclusions[28]. Direct viral damage to the liver is not considered to be the main culprit when multiorgan problems such cardiopulmonary insufficiency, renal impairment, systemic inflammatory status, and the use of several medicines are taken into account. Consequently, the mechanism of liver damage by SARS-CoV-2 infection remains unknown.
SYSTEMIC INFLAMMATION-RELATED LIVER DYSFUNCTION IN COVID-19
Although the majority of COVID-19 patients experience a moderate early disease onset, some patients' conditions quickly worsen and they may experience multiple organ failure as a result of an inflammatory "cytokine storm"[29,30]. Tumor necrosis factor-α, interleukin(IL)-2, IL-6, IL-7, IL-18, granulocytecolony stimulating factor, interferon-γ, monocyte chemotactic protein 1, macrophage inflammatory protein 1 alpha, interferon-inducible protein-10, and ferritin are examples of inflammatory cytokines that are exuberantly released during an inflammatory cytokine storm[9,30]. Patients with severe disease had significantly higher peripheral blood levels of the aforementioned variables than patients with mild disease[31,32]. Hypercytokinemia that is deadly or fulminant may set off a series of events that damage the liver[33]. In patients with COVID-19, lymphopenia and high C-reactive protein levels may operate as independent predictors of hepatic damage, which may be caused by an inflammatory cytokine storm[34]. Through the activation of killer T cells and toll-like receptors (TLRs), SARS-CoV-2 can directly produce a number of proinflammatory signals[35]. Following SARS-CoV-2 infection, activated T lymphocytes attack the infected cells, causing them to die and become necrotic until there are no more T lymphocytes left. TLRs, chemicals associated with damage, that are generated by infected dead cells, can intensify the inflammatory signals. Failure to control viral and bacterial infections caused by Tlymphocyte depletion results in the activation of various inflammatory signaling pathways that activate macrophages thereby causing subsequent inflammatory reactions. In addition to the lungs, this vicious cycle can harm numerous organs, including the liver. Furthermore, it has been observed that COVID-19 patients show hepatic impairment due to a severe cytokine storm rather than the direct cytopathogenic effects of SARS-CoV-2 itself[36-39]. Patients with COVID-19 are more likely to have a number of hepatic illnesses, such as non-alcoholic fatty liver disease, liver cirrhosis, hepatocellular carcinoma, hepatitis B, and hepatitis C[36,40,41].
HYPER-FERRITINEMIA IN COVID-19 AND LIVER INJURY
It has been established that hyperferritinemia is a distinctive symptom of severe COVID-19[42]. The severity and poor prognosis of patients with COVID-19 have been directly associated with ferritin levels[33]. Although the liver significantly contributes to the levels of circulating serum ferritin in COVID-19, proximal tubule cells of the kidney and splenic macrophages are also two potential biological sources. Hepatocytes not only actively secrete ferritin[43], but also release ferritin after hepatic cell death[44]. Furthermore, by producing iron in enterocytes and macrophages, the important iron-regulatory hormone hepcidin may raise intracellular ferritin levels[45]. Majority of patients had inflammationdependent elevation of hepcidin levels to varying degrees in severe illnesses with hyperinflammation[46]. The severity of COVID-19 is associated with elevated serum hepcidin levels[46,47]. Interestingly, SARS-CoV-2 can imitate hepcidin without triggering an inflammatory response by causing ferroportin blockage, which results in high ferritin levels[48].
IRON OVERLOAD IN LIVER INJURY CAUSED BY COVID-19: INTERMEDIATION OF FERRITINOPHAGY
The body's main organ for storing iron is the liver. According to growing evidence, lytic cell death processes like necroptosis, pyroptosis, and ferroptosis cause intense inflammatory reactions by releasing cellular components and permeating cell membranes. This results in the activation of hepatic stellate cells (HSCs) and the recruitment of immune cells[49]. It is without dispute that both hereditary and acquired iron in excess contribute to liver damage. Due to the active mobilization of cellular iron caused by the stimulation of ferritinophagy, ferroptosis may be used to evaluate excess iron. However, unrestrained free iron is harmful to the liver, promoting the development of hepatic disorders and producing serious side effects[49].
The most important cellular iron storing protein is ferritin. The liver is the primary organ for storing iron as a ferritin complex and plays a crucial role in maintaining iron homeostasis. It has been proven that excessive iron conditions may lead to liver damage. Different types of liver disorders are largely caused by iron-catalyzed oxidative damage[50,51]. The release of free iron from the broken hemoglobin and ferritin catabolism may cause COVID-19-related iron excess. High blood levels of free iron may result from ferritin, losing some of its internal iron content[52]. Ferritin abundance is a major determinant of iron homeostasis, as proved by the fact that iron deposits produced by ferritin result in a weakly labile iron pool. Contrarily, the release of iron into the labile iron pool, as a result of ferritin depletion, increases vulnerability to ferroptosis[53]. Studies have hypothesized that ferroptosis in fibroblasts and cancer cells is influenced by the selective autophagic turnover of ferritin (ferritinophagy)[54]. Ferritin's cargo receptor, nuclear receptor coactivator 4 (NCOA4), attaches and transports it to autophagosomes for ferritin breakdown and iron release[55,56]. Ferritinophagy has previously been shown to have important roles in the pathological processes of neurodegeneration, cancer, ischemia/reperfusion injury, and urinary tract infections[57], however, its possible involvement in COVID-19 remains unknown[58]. For the past 4 years, ferritinophagy has been involved in physiology and pathology process of liver, including hepatic insulin resistance[59], hepatocyte senescence[60], ferroptosis in hepatic stellate cells[61-63], hepatocellular carcinoma[64,65] and liver fibrosis[66] (Supplementary Table 1).
In COVID-19 patients, iron depletion or chelation has been suggested as a possible antiviral treatment to guard against severe inflammatory reactions and tissue damage by sequestering iron and inhibiting the generation of oxygen radicals and lipid peroxidation[67]. Poonkuzhiet al[68] mentioned that deferasirox administered orally, along with intravenous deferoxamine made iron chelation therapy effective for COVID-19 victims. A case-control study showed that iron chelators which reducd iron intake could be considered a therapeutic goal of COVID-19[69]. Additionally, the iron chelator and lactoferrin can block SARS-CoV-2 receptor binding for entrance into host cells[70,71].
FERRITINOPHAGY-MEDIATED FERROPTOSIS IN LIVER INJURY CAUSED BY COVID-19
Excess intracellular iron can produce reactive oxygen species (via Haber-Weiss and Fenton processes), reactive nitrogen species, and reactive sulfur species by reacting with molecular oxygen[72]. Redox injury favors mitochondrial malfunction, leading to ferroptosis, various tissue damages, and eventual fibrosis[73]. A new genetically encoded form of programmed cell death called ferroptosis is caused by iron-dependent lipid peroxidation[74]. Opportunities for diagnosis and treatment have been created by further functional understanding of ferroptosis' role in liver fibrosis development[74,75]. Artesunate reduces liver fibrosis by modulating the ferroptosis signaling system, according to a recent publication by Konget al[66]. Additionally, Suiet al[76] demonstrated that by controlling the ferroptosis signaling pathway, magnesium isoglycyrrhizinate reduces liver fibrosis and HSC activation. Moreover, for artemether to reduce liver fibrosis and HSC activation brought on by carbon tetrachloride, p53-dependent induction of ferroptosis is required[77]. For artemether to effectively treat hepatic fibrosisviathe ferroptosis pathway, iron regulatory protein 2 is necessary. Moreover, by inhibiting lipid peroxidation and glutathione depletion, ferrostatin-1, deferoxamine, and vitamin E may have a protective impact on hepatocytes[78].
Iron is abundant in HSCs, which is necessary for ferroptotic cell death[62]. Presumably, ferroptosis encourages the onset and progression of liver fibrosis. It has been found that liver fibrosis caused by acetaminophen in mice is amplified by excessive hepatic iron deposition and ferroptosis, which can be reversed by ferrostatin-1[79,80]. Ferroptosis was also shown by Zhouet al[81-83] to be a kind of autophagy-dependent cell death. Numerous studies have suggested that autophagy controls cellular iron homeostasis and the production of reactive oxygen species, thereby acting as an upstream mechanism in the activation of ferroptosis[81-83]. According to Wanget al[84], ferric citrate, a ferroptosis stimulant, potently causes ferroptosis in murine primary hepatocytes and bone marrowderived macrophages, which prevents the healing of liver injury. Chenet al[85] proposed that ferroptosis caused by reactive oxygen species contributes to liver damage caused by COVID-19.
Notably, a number of recent investigations have shed light on controlled cell death and underlined the significance of autophagy as an emerging mechanism of ferroptosis[86,87]. Multiple routes, including ferritinophagy, which is NCOA4 dependent, may be involved in the molecular mechanisms[54]. Thus, during ferroptosis, ferritinophagy promotes iron accumulation and free radical damage. Dihydroartemisinin, according to Duet al[88], reduces leukemia cells' ability to proliferate and causes ferroptosis by degrading ferritin under the control of autophagy. Moreover, Konget al[66] demonstrated that artesunate reduced HSC ferroptosis caused by ferritinophagy. Notably, the activation of ferritinophagy is required for the RNA-binding protein, embryonic lethal vision-like protein 1, to regulate ferroptosis in HSCs[62]. New diagnostic and therapeutic ways to control HSC survival and death in liver fibrosis may be provided by further investigations of the post-transcriptional regulating mechanisms of ferroptosis. However, as hepatocytes die, ferritin is further released. Due to the mutual stimulation of ferritin and hepatocyte destruction, a vicious cycle is created that continuously worsens liver damage.
CONCLUSION
In light of this, we speculate that cytokine storms caused by SARS-COV-2 infection can encourage hyper-ferritinemia, exacerbating inflammation. Elevated ferritin levels can cause ferroptosis, cell death, and liver damage by inducing NCOA4-mediated ferritinophagy. Therefore, in COVID-19 patients, ferroptosis can be addressed to reduce liver damage and disease severity by limiting ferroptosis (Figure 1). To validate the role of ferritinophagy-mediated ferroptosis in SARS-CoV-2 infection, further research is required. Drug designing to target hepatic cells ferritinophagy-mediated ferroptosis would be a novel approach in the treatment of COVID-19-induced liver injury.
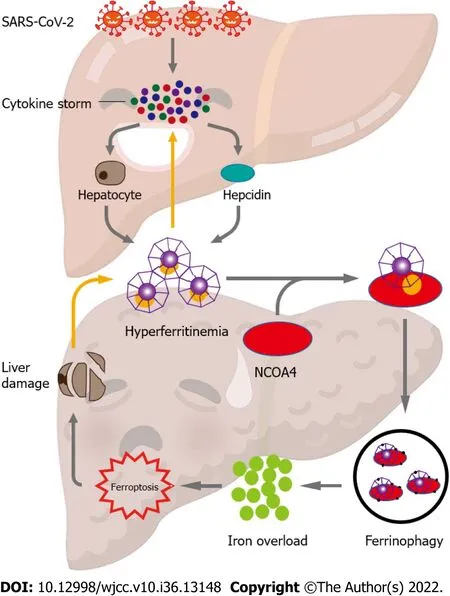
Figure 1 Proposed mechanism of ferritinophagy-mediated ferroptosis in severe acute respiratory syndrome coronavirus 2 infectioninduced liver injury. High levels of inflammation characterized by cytokine storms are caused by severe acute respiratory syndrome coronavirus 2 infection.These cytokine storms cause hyper-ferritinemia by stimulating hepatocytes to secrete ferritin and upregulate hepcidin levels, which further amplifies inflammation. The nuclear receptor coactivator 4 binds to ferritin and delivers it to autophagosomes for ferritin degradation and iron release. Ferroptosis is generated by the excess of intracellular iron, consequently resulting in liver injury. The death of hepatocytes further releases ferritin. Thus, the mutual promotion of ferritin and hepatocyte damage generates a vicious loop that constantly heightens liver injury.
FOOTNOTES
Author contributions:Jia FJ designed and wrote the manuscript; Han J revised the manuscript; All authors have read and approve the final manuscript.
Supported byShandong Provincial Natural Science Foundation, No. ZR2020QC088.
Conflict-of-interest statement:All the Author report no relevant conflicts of interest for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non
commercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORCID number:Feng-Ju Jia 0000-0002-5406-913X.
S-Editor:Liu JH
L-Editor:A
P-Editor:Liu JH
 World Journal of Clinical Cases2022年36期
World Journal of Clinical Cases2022年36期
- World Journal of Clinical Cases的其它文章
- Amebic liver abscess by Entamoeba histolytica
- Living with liver disease in the era of COVID-19-the impact of the epidemic and the threat to high-risk populations
- Cortical bone trajectory screws in the treatment of lumbar degenerative disc disease in patients with osteoporosis
- Probiotics for preventing gestational diabetes in overweight or obese pregnant women: A review
- Effectiveness of microwave endometrial ablation combined with hysteroscopic transcervical resection in treating submucous uterine myomas
- Antibody and complement levels in patients with hypersplenism associated with cirrhotic portal hypertension and therapeutic principles