
不同色谱技术测定乳制品中N-乙酰神经氨酸的比较研究
2022-12-30 06:12宋戈刘九阳苗晶李博群
中国乳品工业 2022年12期
宋戈,刘九阳,苗晶,李博群
(1.黑龙江省绿色食品科学研究院,哈尔滨 150028;2.东北农业大学食品学院,哈尔滨 150030)
0 引言
N-乙酰神经氨酸广泛存在于多种生物组织中,是构成细胞膜上糖蛋白和糖脂的重要成分,在大部分哺乳动物组织中发现的唾液酸主要是N-乙酰神经氨酸,所以通常把N-乙酰神经氨酸称为唾液酸,它参与细胞表面的多种生理功能。另外,N-乙酰神经氨酸是一种天然的大脑营养素,它能促进婴儿的记忆力和智力发育[1]。N-乙酰神经氨酸的食物来源主要是母乳,尤其是初乳中的含量最高,也存在于牛奶、鸡蛋和奶酪中。随着国家卫生计生委发布2017年7号公告,N-乙酰神经氨酸已作为营养素添加到除婴幼儿食品外的食品中,尤其是调制配方粉。分析N-乙酰神经氨酸的方法很多,主要有高效液相色谱法[2-10]、气相色谱法[11]、高效液相色谱串联质谱法[12-14]、脉冲电化学高效阴离子交换色谱检测法[15-18]等。
本研究尝试建立离子交换色谱测定乳制品中N-乙酰神经氨酸的方法,优化水解提取的条件和离子色谱分析条件。比较离子交换色谱法、超高效液相色谱串联质谱法和液相色谱法测定乳制品中N-乙酰神经氨酸后,采用离子交换色谱法测定N-乙酰神经氨酸,操作简便,结果可靠,干扰较小,可为乳制品中N-乙酰神经氨酸含量的测定提供参考。
1 材料与方法
1.1 材料与试剂
N-乙酰神经氨酸(≥99%),德国Dr.Ehretorfer公司;50%氢氧化钠(色谱纯)、乙酸钠(色谱纯)、乙酸铵(色谱纯)、甲酸(色谱纯),SIGMA公司;浓硫酸(分析纯)、氢氧化钠(分析纯),西陇公司;乙腈(色谱纯)、四氢呋喃(色谱纯),美国默克公司;磷酸(色谱纯),科密欧公司;所有用水均为电阻率≥18.2 MΩ·cm的超纯水;以上试剂除标注的外均为分析纯。
1.2 仪器与设备
AB4500超高效液相色谱串联质谱仪,美国AB公司;ICS-5000离子色谱仪配脉冲安培检测器,thermo公司;LC-20A高效液相色谱仪配荧光检测器,日本岛津公司;TTL-DCⅡ水浴加热仪,北京同泰联科技有限公司;pB-10酸度计,德国Sartorius公司;KQ-250DE超声波振荡器,昆山市超声仪器公司;VORTEX3涡旋混合器,德国IKA公司;SQP天平,德国Sartorius公司。
1.3 实验方法
1.3.1 标准溶液与淋洗液的配制
N-乙酰神经氨酸标准储备液(500 mg/L):准确称取0.05 g(精确至0.1 mg)的N-乙酰神经氨酸标准物质于25 mL容量瓶中,加入约10 mL水,超声溶解后冷却至室温,用水定容、摇匀。
N-乙酰神经氨酸标准工作溶液:取适量N-乙酰神经氨酸标准储备液,用水配制成质量浓度为1.0、5.0、10.0、25.0、50.0、100.0、1 000.0 mg/L的标准曲线工作溶液,用于离子色谱法和液相色谱法测定。取适量10.00 mg/L的N-乙酰神经氨酸标准工作溶液,用乙腈 配 制 成 质 量 浓 度 为5.0、10.0、50.0、100.0、200.0、500.0 μg/L的标准曲线工作溶液,用于超高效液相色谱串联质谱法测定。
淋洗液A纯水:电阻率≥18.2 MΩ·cm的超纯水。
淋洗液B氢氧化钠溶液(200 mmol/L):取10.4 mL 50%氢氧化钠溶液,用水稀释至1 000 mL,通入氮气保护,缓慢摇匀。
淋洗液C乙酸钠(500 mmol/L)混合溶液:称取41 g无水乙酸钠(精确至0.01 g),用约500 mL水溶解,0.22 μm滤膜过滤,脱气10 min,加入7.8 mL 50%氢氧化钠溶液,并用水稀释至1 000 mL,通入氮气保护,缓慢摇匀。
1.3.2 样品处理
称取调制乳粉或配方乳粉约10.0 g于50 mL三角瓶中,加入50 mL(50±5℃)的水将试样振荡溶解并超声20 min;称取液奶或酸奶约50 g,用水转移定容至100 mL摇匀。准确移取10 mL定容摇匀的溶液,加入10 mL 0.1 mol/L硫酸,80度水浴1 h,取出冷却用1.0 mol/L NaOH溶液调节pH到4.5~4.6后转移,用水定容到25 mL,离心过滤,滤液备用。
1.3.3 离子交换色谱法测定
色谱柱:CarboPac PA10,250×4.6 mm;保护柱:CarboPac PA10,50×4 mm;检测器参数:Au电极,AgCl参比模式,标准四电位波形;流速:1.0 mL/min。
淋洗条件:0~15 min,120 mmol/L NaOH 60 mmol/L NaAc(50%淋洗液B,12%淋洗液C);15.1~25 min,100 mmol/L NaOH 500 mmol/L NaAc(100%淋洗液C);25.1~35 min,200 mmol/L NaOH(100%淋洗液B);35.1~45 min,120 mmol/L NaOH 60 mmol/L NaAc(50%淋洗液B,12%淋洗液C)。进样方式:自动进样;进样体积:25 μL;柱温:30℃。
取备用滤液和标准工作液过膜上机测定。
1.3.4 超高效液相色谱串联质谱法测定
色谱柱:Waters HILIC 100×2.1 mm,1.7 μm;流动相:A10 mmol/L乙酸铵溶液,B乙腈。
洗脱梯度:0~1.0 min,95% B;1.0~2.0 min,95% B~50%B;2.0~5.0min,50%B;5.0~6.0 min,50% B~95%B;6.0~9.0 min,95%B。进样方式:自动进样;进样量:5 μL;柱温:30℃;流速:400 μL/min。
质谱参考条件:电离方式,电喷雾正离子模式;检测方式,多反应监测(MRM);离子源温度(TEM):550℃;雾化气(Gas1):45;辅助气(Gas2):55;气帘气(Gurtain Gas):30;电 喷 雾 电 压:5 500 V;碰 撞 气(CAD):12;定性离子、定量离子、碎裂电压和碰撞能量见表1。

表1 化合物定性、定量离子和质谱分析参数
移取备用滤液1 mL于10 mL容量瓶中,用乙腈定容摇匀过膜后和标准工作液直接上机测定。
1.3.5 液相色谱法测定
色谱柱:岛津AQ-C18,250×4.6 mm,5 μm;检测器参数:荧光检测器,激发波长230 nm,发射波长425 nm;流速:1.0 mL/min。流动相:95%乙腈水溶液+1%四氢呋喃水溶液(含0.2%磷酸)=9∶1;进样方式:自动进样;进样体积:25 μL;柱温:30℃。
分别取备用滤液和系列标准工作液1.0 mL,于15 mL离心管中,再加入1.0 mL浓度为5 mg/mL邻苯二胺溶液(用0.1 mol/L硫酸氢钠配制),摇匀后置于80℃水浴下衍生50 min,冷却,过膜上机测定。
2 结果与讨论
2.1 水解条件的优化
N-乙酰神经氨酸是一种碳水化合物,它以九碳酮糖酸-神经氨酸为骨架,通常在糖蛋白或糖脂的末端以糖苷的形式存在[1,19]。乳制品中的N-乙酰神经氨酸主要以3种形式存在:游离态、蛋白结合态以及低聚糖结合态。为准确测定乳制品中N-乙酰神经氨酸的含量,需要释放出结合态的N-乙酰神经氨酸。依据文献报道[2-4]选取0.1 mol/L硫酸在80℃的温度条件下水解条件,以尽可能地完全水解释放N-乙酰神经氨酸,对含N-乙酰神经氨酸的乳粉样品及加标样品进行不同水解条件的测试。考察了在80℃的温度条件下,当加入10 mL 0.1 mol/L硫酸溶液,不同反应时间对测定结果的影响如图1所示;考察了当反应时间为60 min,加入不同体积的0.1 mol/L硫酸溶液对测定结果的影响如图2所示。最终确定为1.3.2中的水解条件:10 mL样品溶液加入10 mL、0.1 mol/L硫酸溶液,在80℃水浴60 min水解,用离子交换色谱法进行测定,结果较为稳定,回收率较好。

图1 80℃下加入10 mL0.1 mol/L硫酸溶液水解反应时间与回收率

图2 80℃下水解反应60 min加入0.1 mol/L硫酸溶液体积与回收率
2.2 离子色谱法色谱条件的确定
N-乙酰神经氨酸是一种酸性化合物,在碱性淋洗液条件下易变成阴离子,所以N-乙酰神经氨酸在阴离子交换柱上的保留比样品溶液中单糖、双糖都强很多。结合实验室的条件,选取色谱柱容量一般的阴离子交换柱CarboPacPA10,虽然N-乙酰神经氨酸与乳糖很容易分离,但N-乙酰神经氨酸在强碱性条件下容易发生结构改变或取代基移位,所以不能只通过增大NaOH浓度来达到使N-乙酰神经氨酸在阴离子交换色谱柱上尽快洗脱的目的[16],洗脱开始采用120 mmol/LNaOH和60 mmol/LNaAc等度淋洗15 min的有效淋洗,使N-乙酰神经氨酸在尽可能短的时间内得到理想分离。另外,乳制品成分复杂,酸化后的溶液还常常伴随着更难洗脱的低聚糖和糖蛋白,如果没能洗脱下来,会对下一个样品的测定产生影响,导致保留时间的不稳定和测定结果的重复性差,必须采用具有更强淋洗能力的溶液进行洗脱。待N-乙酰神经氨酸洗脱出来后,采用大浓度500 mmol/L NaAc溶液和高浓度200 mmol/L NaOH溶液交替冲洗色谱柱,最后再回到初始浓度的NaAc溶液和NaOH溶液平衡,准备运行下一个样品,最终确定为1.3.3中的淋洗条件。样品中N-乙酰神经氨酸测定结果的稳定性和准确性较好,标样和样品测定色谱如图3和图4所示。
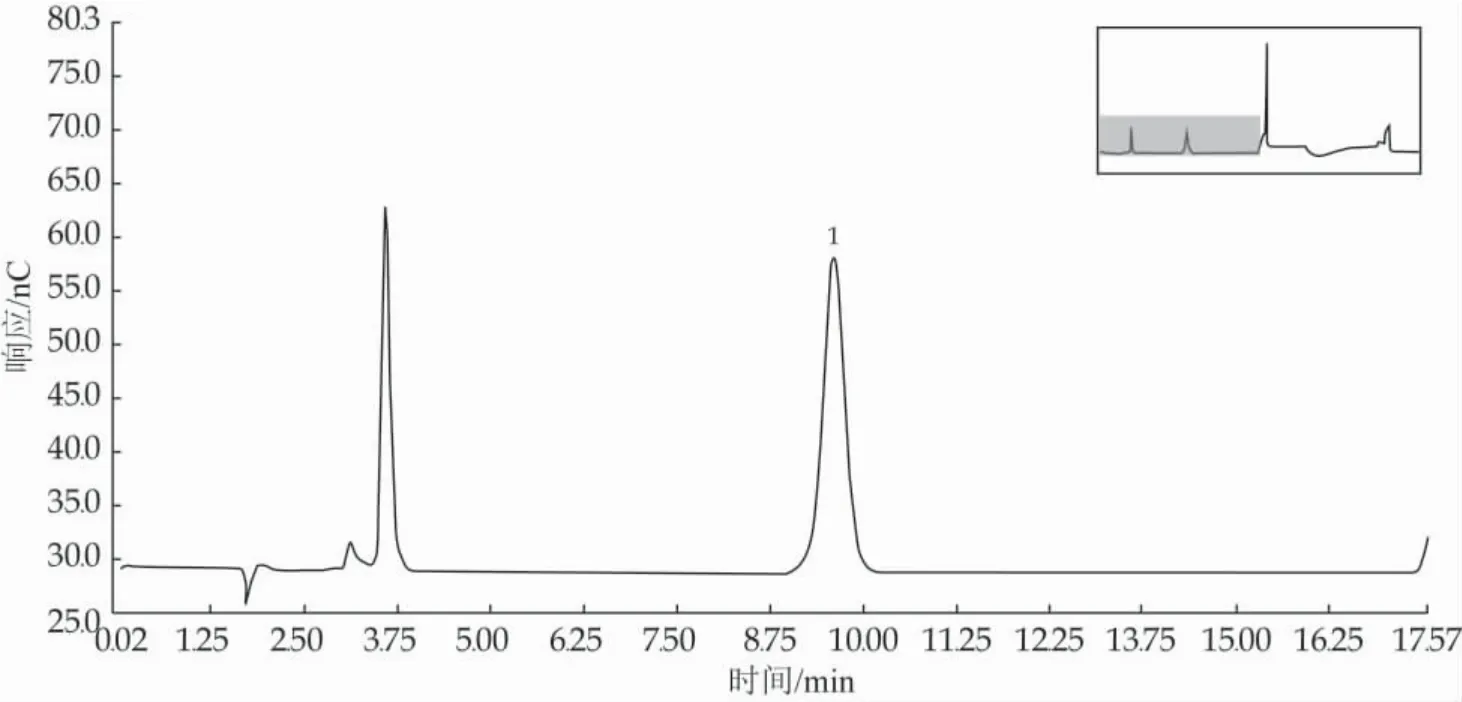
图3 标样测定色谱

图4 配方粉样品测定色谱
2.3 超高效液相色谱串联质谱法条件的确定
在ESI+和ESI-模式下对N-乙酰神经氨酸标准溶液母离子进行全扫描,结果表明在正离子模式下较理想[14],N-乙酰神经氨酸正离子模式下的响应或灵敏度比负离子模式[13]高出一倍。在ESI+模式下,对N-乙酰神经氨酸的标准溶液进行母离子全扫描,以确定N-乙酰神经氨酸的准分子离子并优化质谱条件。以其准分子离子为其母离子,对其子离子进行全扫描,选择碎片离子丰度较高的两个离子分别作为定性和定量离子(响应值高的子离子为定量离子,另外一个为定性离子),在ESI+和MRM模式下优化各种质谱调谐参数见表1。N-乙酰神经氨酸是一种极性酸性化合物,在一般的键合C18色谱柱上保留很弱或几乎不保留。为了增强保留,可以在流动相中添加阳离子对试剂[12],但对质谱仪会造成污染,甚至会抑制目标物的电离。结合实验室条件,选择N-乙酰神经氨酸有特殊保留的HILIC色谱柱[14]和经特殊处理有氢键作用的C18色谱柱T3[13]进行分离分析比对。同一浓度的N-乙酰神经氨酸经色谱柱分离后在质谱上的响应或灵敏度相差无几,但HILIC色谱柱比色谱柱T3的保留和分离效果更好,如图5和图6所示。

图5 HILIC色谱柱分离选择子离子流色谱

图6 T3色谱柱分离选择子离子流色谱
2.4 液相色谱法探讨
N-乙酰神经氨酸无紫外吸收,但在酸性条件下可与邻苯二氨反应,生成有强紫外吸收及荧光的衍生物[20-24],可以用液相色谱仪荧光检测器检测。依据文献[4]对系列标准工作液和样品溶液进行衍生测定,不同浓度的标准工作液叠加色谱和标准工作液与样品溶液叠加色谱如图7和图8所示。从不同浓度的标准工作液叠加色谱图中可以看出标样衍生后的标志物有不少,而且均呈比例,相互间分离度较差,不利于准确定性和定量计算。

图8 标准工作液与样品溶液叠加色谱
2.5 标准曲线和检出限
取不同浓度1.0、5.0、10.0、25.0、50.0、100.0、1 000.0 mg/L的N-乙酰神经氨酸标准工作溶液,用离子交换色谱仪测定,记录目标峰峰面积。取不同浓度5.0、10.0、50.0、100.0、200.0、500.0 μg/L的N-乙酰神经氨酸标准工作溶液,用超高效液相色谱串联质谱仪测定,记录目标峰峰面积。以峰面积与标准溶液浓度绘制线性回归曲线,并以3倍信噪比计算出固体样品称取10.0 g的检出限,结果见表2。

表2 不同方法的线性和检出限
2.6 回收率和精密度
以同一个全脂乳粉为实验材料,称取10 g样品后,再分别加入1.0 mg和5.0 mg的N-乙酰神经氨酸标样,对样品及两个不同浓度的加标样品进行6次平行测定,计算样品的精密度、回收率及回收率的精密度(RSD)。离子交换色谱法和超高效液相色谱串联质谱法的回收率和精密度见表3。由此可以看出,离子交换色谱法测定结果较为稳定,重复性和准确性较好。

表3 不同方法的回收率和精密度 (n=6)
2.7 实际样品的测定
按照本方法对市售的乳制品样品进行N-乙酰神经氨酸含量的测定,结果见表4。从实际样品的检测结果来看,离子交换色谱法的灵敏度和准确度可以满足实际测试的需求。

表4 不同方法测定样品中N-乙酰神经氨酸的含量mg/100 g
3 结论
本文重点探讨了乳制品中N-乙酰神经氨酸水解的条件,建立了检测N-乙酰神经氨酸的离子交换色谱法。比较了离子交换色谱法、超高效液相色谱串联质谱法、液相色谱法测定N-乙酰神经氨酸的优劣。离子交换色谱法具有良好的线性,操作简便,结果可靠,干扰小,同时重复性好,准确度高,适合乳制品中N-乙酰神经氨酸含量的生产监测和监管检测。
猜你喜欢
分子催化(2022年1期)2022-11-02
中国乳品工业(2022年8期)2022-08-28
当代水产(2022年4期)2022-06-05
中国典型病例大全(2022年9期)2022-04-19
茶业通报(2021年4期)2022-01-21
口腔护理用品工业(2021年4期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
现代牧业(2021年4期)2021-04-14
核农学报(2020年7期)2020-07-01
