
固体超强酸S2/Fe2O3-ZrO2脱除模拟汽油中碱性氮的工艺研究*
2022-12-17 10:18姚凯伦陈大为李树兵
化学工程师 2022年11期
姚凯伦,闫 锋,陈大为,李树兵
(1.辽宁石油化工大学 石油化工学院,辽宁 抚顺 113001;2.中国石油 天然气股份有限公司 抚顺石化分公司 乙烯化工厂,辽宁 抚顺 113001;3.盘锦辽河油田双兴实业有限责任公司,辽宁 盘锦 124000)
现代社会车辆越来越多,油品消耗量增加。油品中含氮化合物燃烧可以产生NOx等有害气体,是重要的大气污染源,同时也会产生酸雨和光化学烟雾等一系列环境问题[1]。氮化物不仅会使催化剂活性中心减少,造成催化剂中毒,而且会使油品的安定性变差[2],脱除碱性氮化物有重大意义。
氧化脱氮是被广泛研究的一种方法。固体超强酸是指酸性超过100%H2SO4的固体酸[3]。在有机合成、有机物降解、烷烃异构化和催化氧化脱硫等方面有诸多研究[4,5]。传统液体酸催化剂会有一部分溶解在油品中降低油品的品质,固体酸在反应后容易回收、再生且不影响油品品质。
1 反应机理
1.1 酸中心形成机理
S2型固体超强酸的酸中心是通过S2与金属氧化物表面的吸附配位作用形成的。有报道[7]称在固体超强酸中S=O键为共价键,S和O的电负性不同,导致电子云发生偏移从而产生超强酸位,使金属附近出现较强的L酸中心,形成固体超强酸。
1.2 氧化原理
固体超强酸的酸中心能与H2O2反应形成氧化性更强的过氧超强酸,在反应时把含氮化合物氧化为氮氧化物,氮氧化物和酸生成铵盐从而被体系中生成的水萃取脱除[8]。
2 实验部分
2.1 药品及仪器
30%H2O2(AR莱阳经济技术开发区精细化工厂);25%NH3·H2O(沈阳试剂三厂);正辛烷(AR天津市大茂化学试剂厂);喹啉、Zr(NO3)4·5H2O、Fe(NO3)3·9H2O、(NH4)2S2O8均为分析纯,天津市大茂化学试剂厂。
101A-1型恒温干燥箱(上海实验仪器厂);85-2型磁力恒温搅拌器(金坛区华城润华实验仪器厂);SX2-12-10型箱式电阻炉(苏州江东精密仪器有限公司);TSN-5000型紫外荧光硫氮分析仪(江分电化学仪器有限公司)。
2.2 催化剂的制备
按 物 质 的 量 比4∶1称取Fe(NO3)3·9H2O和Zr(NO3)4·5H2O分别溶解在适量蒸馏水中,缓慢滴加NH3·H2O调节pH值为9~10。室温陈化12h后过滤,将沉淀放至烘箱中干燥。将干燥后的固体研磨后按照1g/15mL的比例加入到0.5mol·L-1的(NH4)2S2O8溶液中,浸渍一定时间后过滤,并在120℃下干燥,在600℃下焙烧3h后得到催化剂。
模拟油的配制 用1mL移液管移取0.4mL喹啉,加入到500mL正辛烷中,摇匀后过夜备用。
2.3 模拟油氧化脱除碱性氮化物
待温度升到设定值后,把反应器置于恒温磁力搅拌器中,取15mL模拟油、适量催化剂和H2O2加入反应器中,在磁力搅拌下反应一段时间。反应结束后过滤,取上层油样,用紫外荧光硫氮分析仪测定反应后模拟油中碱性氮化物含量。过滤出的催化剂用乙醇清洗后烘干,进行后续催化剂回收测试。
取反应前模拟油用紫外荧光硫氮分析仪测得碱性氮浓度为X1ppm,经反应后的模拟油测得碱性氮浓度为X2ppm,则脱除碱性氮率计算公式:
3 结果与讨论
3.1 催化剂的表征
3.1.1 XRD表征 图1为3种催化剂的XRD谱图。
3.1.2 FT-IR表征 图2中3种催化剂在1634cm-1和3417~3430cm-1处的特征峰为催化剂吸水后产生的O-H键的伸缩振动和弯曲振动[9]。1620~1630cm-1处未发现明显的S=O键的伸缩振动峰,表明不存在一般认为,在1200cm-1以下时,金属氧化物与S2为桥式双配位;在1200cm-1以上时,金属氧化物与S2为螯合双配位[11]。所以,催化剂中S2以桥式双配位和螯合双配位两种形式共存。S=O键具有强烈的吸电子效应,使催化剂中的金属离子的L酸性增强,从而表现出超强酸的性质[12]。

图2 FT-IR谱图Fig.2 FT-IR spectra
3.1.3 BET分析 图3为3种催化剂的吸附脱附等温线为Ⅳ型。
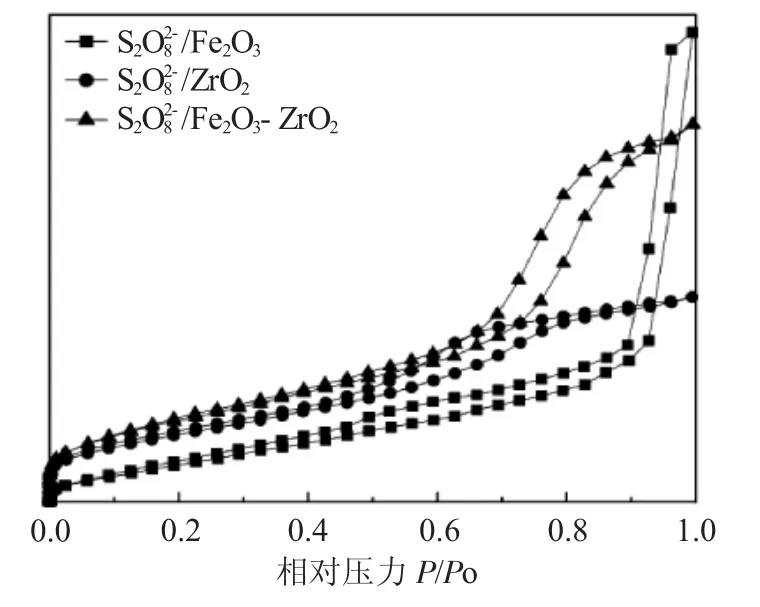
图3 N2吸附/脱附等温线图Fi.3 N2 adsorption-desorption isotherms
由表1可知,经过掺杂,增大了催化剂的比表面积和孔径。比表面积的增加,能够暴露出更多的活性位点,使超强酸位点增多,从而使催化剂有更强的酸性。孔径的增大,有利于扩散过程在孔道中进行,从而提高反应速率。

表1 固体超强酸催化剂的物性参数Tab.1 Surface texture parameter of solid super acid catalyst
3.1.4 NH3-TPD表征 图4为3种催化剂的NH3-TPD曲线图。
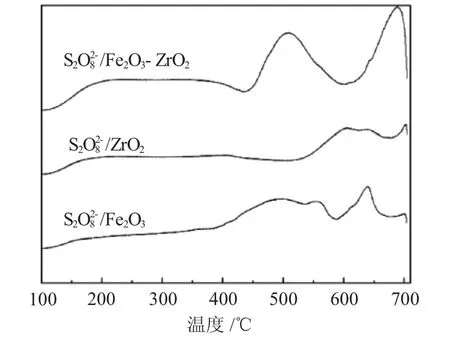
图4 NH3-TPD曲线图Fig.4 NH3-TPD curves
3.2 固体超强酸催化剂在模拟油中脱除碱性氮化物的单一条件考察
3.2.1 催化剂用量对脱碱性氮率的影响 图5为催化剂用量对脱碱性氮率的影响,反应时间1h,反应温度50℃,模拟油用量与氧化剂体积比为10∶1。
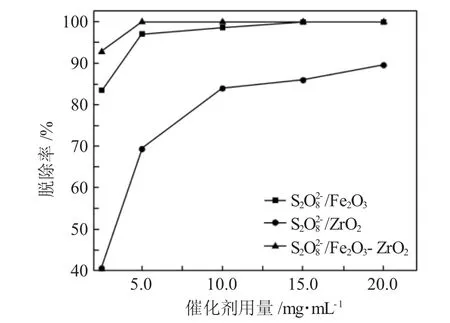
图5 催化剂用量对脱碱性氮率的影响Fig.5 Effect of catalyst dosage on denitrification rate
3.2.2 反应时间对脱碱性氮率的影响 图6为反应时间对脱碱性氮率的影响,催化剂用量为0.005g·mL-1,反应温度为50℃,模拟油用量与氧化剂体积比为10∶1。
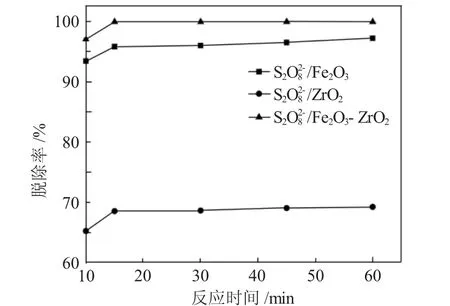
图6 反应时间对脱碱性氮率的影响Fig.6 Influence of reaction time on denitrification rate
3.2.3 反应温度对脱碱性氮率的影响 图7为不同反应温度对脱碱性氮率的影响,催化剂用量为0.005g·mL-1,反应时间为15min,模拟油用量与氧化剂体积比为10∶1。

图7 反应温度对脱碱性氮率的影响Fig.7 Effect of reaction temperature on denitrification rate
由图7可知,随着温度的增加脱除率增加,这可能因为温度升高使过氧超强酸产生的速率增加,同时也增加了过氧超强酸与碱性氮化物反应速率,50℃时催化剂的脱除率最高,达到99%。其与催化剂在温度较低时脱除率有较大差别,这可能是因为引入ZrO2以后ZrO2催化剂的部分活性中心的活化能增加,温度升高后,使这部分活性中心被活化,因而脱除率增加[13]。再升高温度脱除率呈下降趋势,这可能由于每升高10℃,H2O2分解速率提高2~3倍导致的[16]。
3.2.4 氧化剂用量对脱碱性氮率的影响 图8为氧化剂用量对脱碱性氮率的影响,催化剂用量为0.005g·mL-1,反应温度为50℃,反应时间为15min。

图8 氧化剂用量对脱碱性氮率的影响Fig.8 Effect of oxidant dosage on denitrification rate
由图8可知,脱除率随着氧化剂用量的增加而增加,因为随着H2O2用量增加,与催化剂活性中心反应产生的过氧超强酸数量也随之增加。Fe2O3-ZrO2催化剂在氧化剂与模拟油用量体积比为1∶20时,脱除率达到最高,为99%。
3.3 催化剂重复利用次数考察
由图9可知,随着催化剂使用次数的增加,碱性氮的脱除率呈下降趋势。在前3次催化剂循环使用中,脱除率均维持在较高水平。循环次数达到3次后,脱除率随之下降,第四次为82%,循环5次时脱氮率降至54%。这是由于催化剂表面活性组分流失或者氮化物覆盖在催化剂表面,造成酸中心与氧化剂及碱性氮化物接触减少导致的。通过补充活性组分和活化处理能解决此问题。
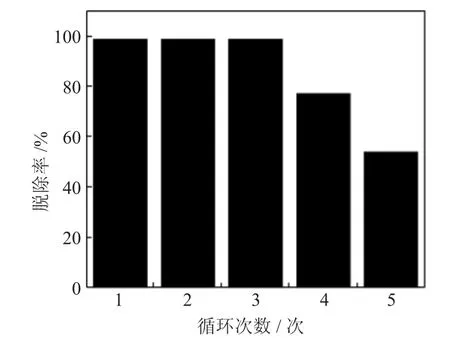
图9 S2/Fe2O3-ZrO2催化剂循环次数对脱碱性氮率的影响Fig.9 Effect of S2/Fe2O3-ZrO2 catalyst cycle timeson the denitrification effect
4 结论
(1)通过XRD、FT-IR、NH3-TPD、BET对催化剂进行表征,结果表明,经过掺杂后催化剂ZrO2的比表面积及孔径增大,酸性较强,酸量增多,在进行脱碱性氮实验中,相比催化效果明显,无论从反应时间、催化剂使用量、氧化剂用量方面都是最小的,脱除率达到99%。
猜你喜欢
石油学报(石油加工)(2022年3期)2022-05-11
石油炼制与化工(2021年5期)2021-05-12
航空制造技术(2020年15期)2020-11-06
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
表面技术(2019年6期)2019-06-26
中国有色金属学报(2018年2期)2018-03-26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
浙江农业学报(2017年1期)2017-05-17
浙江农业科学(2016年11期)2016-05-04
