
右美托咪定通过抑制TXNIP保护大鼠急性心肌缺血损伤
2022-12-16 05:14刘晟楠马姣姣李立萍张勤增解丽君
中国药理学通报 2022年12期
刘晟楠,王 媛,马姣姣,郝 娜,姜 红,李立萍,张勤增,解丽君,
(1. 河北大学临床医学院,河北 保定 071000;2. 承德医学院中药研究所,河北 承德 067000;3. 河北医科大学第一医院内分泌科,河北 石家庄 050030;4. 河北医学科学院药物研究所,河北 石家庄 050021)
缺血性心血管病严重危害人类健康,因而对于缺血心肌保护的研究一直是心血管研究的重点。氧化应激作为心肌缺血损伤的一种重要机制,在心肌缺血损伤早期发挥着重要的作用[1]。硫氧还蛋白相互作用蛋白(thioredoxin interaction protein,TXNIP) /硫氧还蛋白(thioredoxin,TRX)系统是机体内存在的复杂氧化还原调节网络中重要的一种。TRX是一类巯基抗氧化蛋白,具有直接清除自由基和氧化还原功能。TXNIP是TRX的一种内源性抑制蛋白,与TRX活性位点结合可以抑制TRX活性,间接调控细胞氧化还原状态,并且可诱导氧化应激的产生。TXNIP/TRX系统通过抗氧化调节作用参与多种细胞或器官的保护[2]。 硫化氢(hydrogen sulfide,H2S)作为一种新型气体信号分子,本实验室前期研究证实,H2S参与了大鼠急性心肌缺血的病理生理过程,H2S供体可通过抗氧化应激,抑制心肌细胞凋亡减轻心肌缺血损伤[3]。
右美托咪定(dexmedetomidine,DEX)是一种高选择性的α2-肾上腺素受体激动剂,具有一定的镇静、镇痛作用,易于唤醒,且对于呼吸无明显的抑制作用,减轻麻醉风险故而广泛应用于临床。研究表明右美托咪定对心、肺、脑、肾等器官的缺血损伤具有一定的保护作用[4-6],可以通过减轻炎症反应,调控参与氧化应激的多条通路减轻心肌细胞损伤[7]。但其保护机制与心肌缺血时TXNIP/TRX系统变化是否有关尚未明确。因此,本研究通过建立大鼠急性心肌缺血模型,进一步探讨DEX的心肌保护作用, 并从TXNIP/TRX活性调节、内源性H2S含量及凋亡相关蛋白caspase-3、NOX-4的表达等方面探讨其作用机制。
1 材料与方法
1.1 材料
1.1.1实验动物 健康成年SPF级♂SD大鼠60只,体质量(210~250)g,由北京华阜康生物科技股份有限公司提供,许可证号:SCXK(京)2019-0008,适应性饲养大鼠1周。
1.1.2实验仪器及试剂 右美托咪定(江苏恒瑞医药股份有限公司,191029BP);2,3,5-氯化三苯基四氮唑(2,3,5-triphenyltetrazolium chloride,TTC)(德国Sigma公司,T8877-25G);TXNIP兔单克隆抗体(浙江华安生物有限公司,ET1705-72);TRX兔多克隆抗体(美国Proteintech公司,14999-1-AP);NOX-4兔单克隆抗体(英国Abcam公司,ab13303);caspase-3兔多克隆抗体(美国Proteintech公司,19677-1-AP);脂质过氧化物(lipid peroxide,LPO)测试盒(南京建成生物工程研究所,A106-1-2);还原型谷胱甘肽(reduced glutathione,GSH)测定试剂盒(微板法)(南京建成生物工程研究所,A006-2-1);硫氧还蛋白还原酶(TrxR)活性检测试剂盒(北京索莱宝科技有限公司,BC1155);H2S含量检测试剂盒(北京索莱宝科技有限公司,BC2055);Western blot相关试剂购于北京索莱宝科技有限公司;ECL发光液(莫奈生物科技有限公司,PW30401S);通用型SP试剂盒(北京中杉金桥生物技术有限公司,SP-9000);DAB显色试剂盒(北京中杉金桥生物技术有限公司,ZLI-9018);考马斯亮兰法试剂盒(南京建成生物工程研究所,A045-2-2);Powerlab8/s多通道生理记录仪(澳大利亚ADInstruments);低温高速离心机5810R(德国Eppendorf公司);垂直电泳仪、转移电泳仪以及转移电泳槽(美国Bio-Rad公司);全波长酶标仪(美国BioTek公司);全自动显微照相系统(德国LEICA公司)。
1.2 方法
1.2.1动物分组与建模 采用随机数字表法分为5组(n=12):对照组(C组)、心肌缺血模型组(M组)、心肌缺血模型+10 μg·kg-1右美托咪定组(D10组)、心肌缺血模型+25 μg·kg-1右美托咪定组(D25组)、心肌缺血模型+50 μg·kg-1右美托咪定组(D50组)。适应性饲养大鼠1周后,使用1%戊巴比妥钠剂量为50 mg·kg-1腹腔注射进行麻醉,大鼠麻醉后,记录标准Ⅱ导联心电图;进行无创气管插管,呼吸机调节为RR 40~60次/分,VT10~12 mL。对大鼠皮肤消毒,分离肌层,于大鼠左侧3~4肋间开胸,挤出心脏,找到冠状动脉左前降支使用6-0带针缝合线进行结扎建立急性心肌缺血模型。记录心电图的变化,以S-T段较术前抬高0.15 mV以上作为成功建立模型。其中对照组仅穿线不结扎;M组在缺血前30 min腹腔注射3 mL生理盐水;D10组、D25组、D50组在缺血前30 min按4 mg·L-1浓度腹腔注射DEX 10、25、50 μg·kg-1。
1.2.2测定血流动力学指标 缺血3 h剪开颈部皮肤,分离大鼠右侧颈总动脉,做颈动脉插管,连接换能器,并将导管送入左心室,观察大鼠动脉压力变化,记录大鼠平均动脉压(MAP)、左室收缩压(LVSP)、左室舒张末压(LVEDP)、左室内压上升/下降的最大速率(±dp/dtmax),计算左室发展压(LVDP=LVSP-LVEDP)。
1.2.3测定血清中H2S含量 取材前使用1%戊巴比妥钠(50 mg·kg-1)麻醉,从大鼠腹主动脉取血,静置40 min后12 000 r·min-1离心10 min,取血清,酶标仪预热30 min,将酶标仪波长调节至665 nm,按说明书中步骤操作,测得各样品吸光度计算含量。
1.2.4测定心肌组织中胱硫醚-γ-裂解酶(cystathionine-γ-lyase,CSE)活性 取大鼠心尖组织,称质量,按1 ∶10加入50 mmol·L-1磷酸钾缓冲液(pH=6.8),制成匀浆,4 ℃、4 000 r·min-1离心10 min,取上清备用。取0.1 mL心肌组织匀浆,加入在25 mL锥形瓶中央的体积为1 cm3的中央室中,再加入pH为7.4含100 mmol·L-1磷酸钾缓冲液、10 mmol·L-1L-半胱氨酸、2 mmol·L-15’-磷酸吡多醛的反应液0.9 mL,加1%醋酸锌0.5 mL在中央室,加滤纸增加气/液接触面积。在锥形瓶中充氮气30 s,盖上磨砂玻璃盖。37 ℃孵育90 min,加0.5 mL 50%三氯醋酸终止反应,再37 ℃孵育1 h,转移到试管,加3.6 mL蒸馏水,0.5 mL含20 mmol·L-1N,N-二甲基-对苯二胺硫酸盐的7.2 mol·L-1盐酸和0.4 mL含30 mmol·L-1三氯化铁的1.2 mol·L-1盐酸,混匀,室温静置20 min,使用665 nm波长检测吸光度。根据NaHS标准曲线计算溶液中H2S的含量,以在一个单位时间内每毫克组织能够生成H2S的量来表示CSE活性。
1.2.5测定心肌组织中LPO含量 取大鼠心尖组织称质量,研磨,测定样本浓度,酶标仪预热30 min,调节波长至586 nm,按说明书中步骤操作,测得各样品吸光度计算含量。
1.2.6测定心肌组织中GSH含量 取大鼠心尖组织称质量,研磨,测定样本浓度,酶标仪预热30 min,调节波长至405 nm,按说明书中步骤操作,GSH可与二硫代二硝基甲苯反应生成波长为405 nm的黄色化合物,故测得各样品吸光度计算含量。
1.2.7检测心肌组织中TrxR的活性 取大鼠心尖组织称质量,剪碎,每0.1 g加入1 mL提取液冰上研磨,10 000 r·min-1,4 ℃离心10 min,取上清置冰上,酶标仪预热30 min,调节波长至412 nm,按说明书操作,TrxR可以催化TNB的生成,利用2-乙烯吡啶抑制样本中原有的GSH后,按每个样本每分钟生成1 nmol TNB为一个酶活力单位测定样本5 min内的TrxR活力。
1.2.8TTC染色检测心肌梗死面积 每组随机选择6只大鼠麻醉,取心脏,-80 ℃冰冻5 min后切片,在1.5% TTC溶液中37 ℃避光水浴20 min,拍照,使用ImageJ软件计算切片梗死面积。
1.2.9苏木精-伊红( hematoxylin-eosin,HE)染色观察心肌缺血情况 取大鼠心脏组织使用4%多聚甲醛固定,乙醇梯度脱水,常规石蜡包埋,切片,进行HE染色,镜下观察心肌缺血情况。
1.2.10电镜下观察心肌缺血情况 迅速取心尖部组织,以4%戊二醛固定,0.1 mol·L-1二甲砷酸缓冲液冲洗,1%四氧化锇固定,缓冲液冲洗后,丙酮脱水,环氧树脂浸透,包埋,切片,醋酸铀-枸橼酸铅染色,透射电镜下观察心肌超微结构的变化。
1.2.11免疫组织化学染色观察TXNIP、TRX、caspase-3情况 心脏组织4%多聚甲醛固定,梯度乙醇脱水,常规石蜡包埋,切片,烤片,脱蜡水合,pH 6.0枸橼酸钠修复液高压修复7 min,自然冷却40 min,PBS 5 min/次,清洗3次,内源性过氧化物酶阻断剂避光37 ℃反应20 min,PBS 5 min/次,清洗3次,封闭用正常山羊血清工作液避光37 ℃孵育40 min,滴加TXNIP(1 ∶100)、TRX(1 ∶100)、caspase-3(1 ∶100),4 ℃避光孵育过夜,复温30 min,PBS 5min/次,清洗3次,生物素标记山羊抗小鼠/兔IgG避光37 ℃反应20 min,PBS 5 min/次,清洗3次,辣根酶标记链霉卵白素工作液避光37 ℃反应20 min,DAB显色,苏木精染核,脱水,中性树胶封片。
1.2.12采用Western blot法检测心肌组织中TXNIP、TRX、NOX-4、以及caspase-3蛋白表达水平 取大鼠心尖组织剪碎加入裂解液研磨,静置20 min,4 ℃ 12 000 r·min-1,离心20 min取上清分装于-80 ℃保存。使用考马斯亮蓝试剂盒测定蛋白浓度。100 ℃金属浴5 min加热变性蛋白。按蛋白分子所需配置SDS-PAGE凝胶电泳分离目的蛋白,PVDF膜90 V冰浴转膜,5%血清37 ℃封闭1.5 h,加入TXNIP(1 ∶3 500)、TRX(1 ∶3 000)、NOX-4(1 ∶2 000)、caspase-3(1 ∶2 000)抗体4 ℃摇床过夜,TBST洗膜3次,加羊抗兔二抗(1 ∶5 000)室温孵育2 h,TBST洗膜3次后使用ECL发光液进行发光照相存档,使用ImageJ软件分析,以目的蛋白条带灰度值比β-actin条带灰度值反映其表达水平。
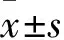
2 结果
2.1 DEX保护心肌缺血大鼠心功能与C组相比,M组大鼠MAP,LVDP,±dp/dtmax均明显减少,LVEDP增加,差异有统计学意义(P<0.01)。与M组相比,D10组、D25组、D50组大鼠MAP,LVDP,±dp/dtmax均增加,LVEDP减少,差异有统计学意义(P<0.05),见Tab 1。
2.2 DEX增加心肌缺血大鼠血清中H2S含量与C组相比,M组大鼠血清中H2S含量明显减少,差异有统计学意义(P<0.01)。与M组相比,D10组、D25组、D50组大鼠血清中H2S含量增加,差异有统计学意义(P<0.05),见Tab 2。
2.3 DEX增加心肌缺血大鼠心肌组织中CSE活性与C组相比,M组大鼠心肌组织中CSE活性明显减少,差异有统计学意义(P<0.01)。与M组相比,D10组、D25组、D50组大鼠心肌组织中CSE活性增加,差异有统计学意义,见Tab 2。
2.4 DEX减少心肌缺血大鼠心肌组织中LPO含量与C组相比,M组大鼠心肌组织中LPO含量明显增加,差异有统计学意义(P<0.01)。与M组相比,D10组、D25组、D50组大鼠心肌组织中LPO含量减少,差异有统计学意义(P<0.01),见Tab 2。
2.5 DEX增加心肌缺血大鼠心肌组织中GSH含量与C组相比,M组大鼠心肌组织中GSH含量明显减少,差异有统计学意义(P<0.01)。与M组相比,D10组、D25组、D50组大鼠心肌组织中GSH含量增加,差异有统计学意义,见Tab 2。
2.6 DEX增加心肌缺血大鼠心肌组织中TrxR的活性与C组相比,M组大鼠心肌组织中TrxR的活性明显减少,差异有统计学意义(P<0.01)。与M组相比,D10组、D25组、D50组大鼠心肌组织中TrxR的活性增加,差异有统计学意义,见Tab 2。

Tab 1 Effect of DEX on cardiac function of myocardial ischemia n=8)
Tab 2 Effects of DEX on H2S,CSE,GSH,LPO and TrxR contents in myocardial ischemia n=6)
2.7 DEX减少心肌缺血大鼠心肌梗死面积与C组相比,M组、D10组、D25组、D50组大鼠心肌梗死面积明显增加,差异有统计学意义(P<0.01)。与M组相比,D10组、D25组、D50组大鼠心肌梗死面积降低,差异有统计学意义(P<0.01),见Fig 1。
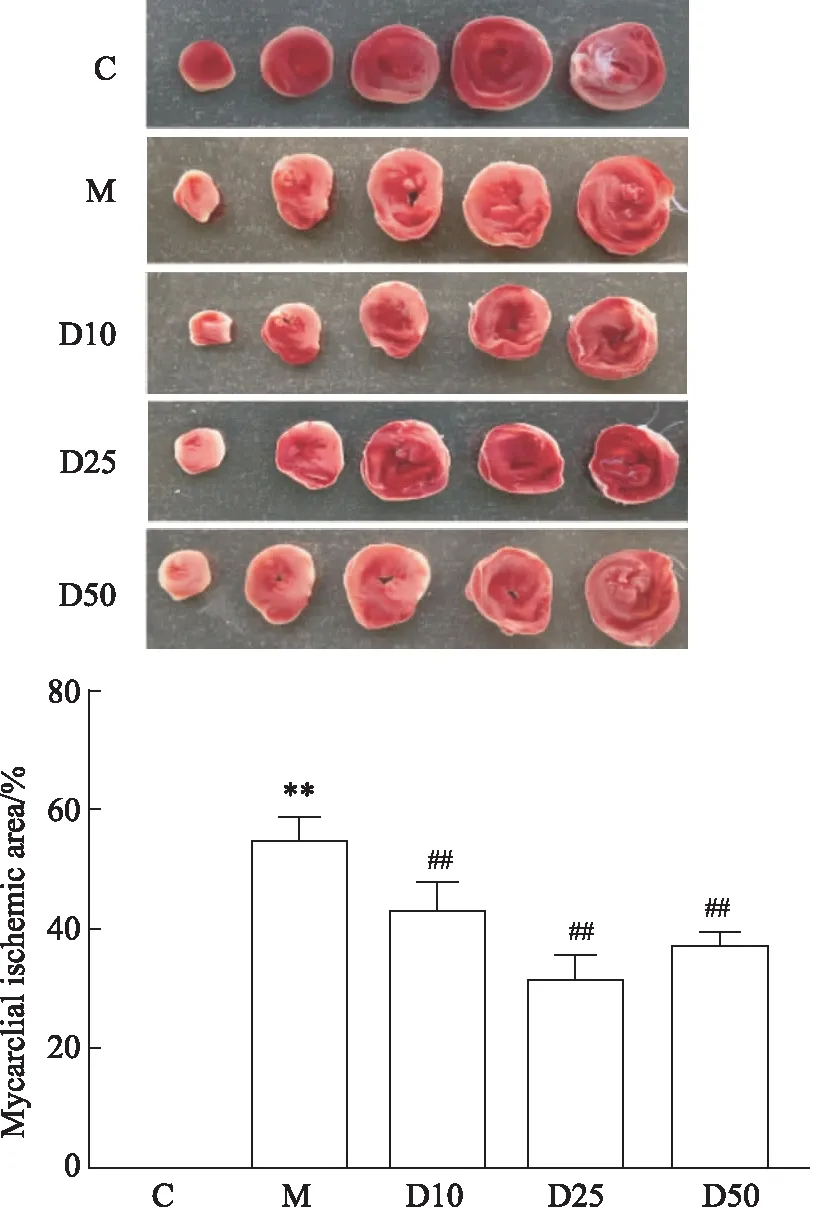
Fig 1 Effect of DEX on myocardial infarction size in myocardial ischemia n=6)
2.8 DEX处理心肌缺血大鼠心肌组织病理变化HE染色结果显示,C组大鼠心肌组织形态结构正常,M组大鼠心肌细胞出现水肿,炎性细胞浸润增加,部分心肌细胞出现裂解,经给药处理后,大鼠心肌细胞形态趋于正常,水肿及裂解细胞减少,炎性细胞浸润减少;电镜下显示M组心肌纤维排列紊乱,核周水肿,核膜局部消失,线粒体嵴膜肿胀变形、溶解、消失,给药组心肌纤维排列紊乱减轻,线粒体嵴膜轻度变形。见Fig 2。
2.9 DEX对心肌缺血大鼠心肌组织中TXNIP、TRX、NOX-4以及caspase-3蛋白表达的影响与C组相比,M组TXNIP、NOX-4、caspase-3表达水平增加,TRX表达水平下降,差异有统计学意义(P<0.05)。与M组相比,D10组、D25组、D50组TXNIP、NOX-4、caspase-3蛋白表达水平下降,TRX表达水平上升,差异有统计学意义(P<0.05),见Fig 3。
3 讨论
随着社会发展和人口老龄化进程加速,心肌缺血发病率逐年增加,且常造成心肌不可逆转的损伤,近年来,心脏疾病患者进行手术的需求也在增加。如何减少心血管疾病患者的手术风险以及合理选择围术期的麻醉药物成为医学研究的热点之一。
心肌缺血损伤是多种机制交互作用(cross-talk)导致的复杂的病理过程,氧化应激在心肌缺血损伤早期发挥着重要作用[1]。心肌缺血时机体处于氧化应激状态,过量氧自由基生成是引起心肌细胞坏死、凋亡或过度自噬和炎症因子释放的主要因素[8]。研究表明一些抗氧化剂是通过抑制氧化应激,调节凋亡和自噬来完成对心肌的保护作用[9]。机体内存在复杂的氧化还原调节网络,其中TXNIP/TRX系统发挥了重要的作用,涉及到多种应激反应的细胞保护机制。TRX系统由TRX、TrxR和NADPH组成,是细胞内关键的硫醇还原系统,通过其二硫化物还原酶活性减轻细胞内氧化应激,维持细胞内蛋白质的还原状态使其发挥正常功能[3]。NADPH的电子通过NOX直接传递给氧分子产生ROS,抑制TRX作用,而NOX-4是NOX中重要的催化亚基[10];TRX还可以与凋亡信号调节激酶1(apoptosis signal-regulating kinase,ASK-1)结合抑制细胞凋亡。TXNIP是TRX的内源性抑制蛋白,TXNIP与TRX活性位点结合而抑制其活性,调节机体的氧化应激状态;TXNIP除间接调控细胞氧化还原状态外,还可诱导氧化应激的产生[2]。有研究表明TXNIP表达上调是糖尿病心肌细胞损伤和凋亡的重要原因,通过控制TXNIP可以减少糖尿病引起的心肌损伤[11],还有研究报道调控TXNIP表达可以减轻肾、小肠等器官的缺血/再灌注损伤[4,12]。
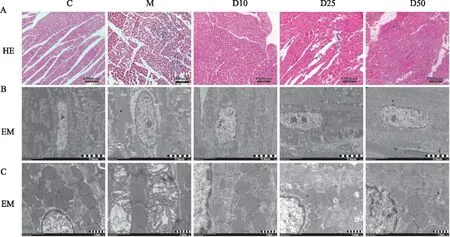
Fig 2 Effect of DEX on histopathology of myocardial ischemia rats
内源性H2S是一种新型的气体信号分子,在心血管系统发挥着重要的生理和病理调节作用。体内H2S主要由L-半胱氨酸在胱硫醚-β-合酶和/或CSE的作用下生成。本实验室前期研究证实,H2S/CSE体系参与了大鼠急性心肌缺血的病理生理过程,H2S供体可通过抗氧化应激,抑制心肌细胞凋亡减轻心肌缺血损伤[3]。MAO等[13]研究表明,内源性H2S可以调节TXNIP/TRX系统,促进TRX与TXNIP解离,提高细胞抗氧化损伤能力,保护肾脏足细胞免于阿霉素所致损伤。在肝细胞,抑制H2S产生的酶CSE活性可以使TRX活性降低,促进TXNIP/TRX的相互作用从而加速细胞的死亡[14]。说明H2S抗氧化活性的发挥与TXNIP/TRX系统密切相关。
DEX作为一种高选择性的α2-肾上腺素受体激动剂,有镇静与镇痛作用,易于唤醒且无呼吸抑制作用,广泛应用于临床。研究表明DEX可以通过PI3K/Akt、mTOR、AMPK/PI3K/Akt/eNOS等信号通路抑制氧化应激,减轻炎症反应起到保护心肌的作用[5,15],但过量DEX也存在降低心率的情况,对心脏的活动有一定的抑制作用而可能增加损伤[16]。本研究在预实验时亦发现,100 μg·kg-1的DEX对大鼠心脏产生明显的负性肌力作用,使得大鼠结扎冠状动脉左前降支术后死亡率升高。
本研究通过结扎大鼠冠状动脉左前降支建立大鼠急性心肌缺血模型,探讨DEX对大鼠急性心肌缺血时内源性H2S和TXNIP/TRX系统的影响,以期进一步明确其可能存在的保护心肌缺血损伤的机制。结果表明,结扎大鼠冠状动脉左前降支造成心肌缺血3 h后,心肌组织CSE活性明显降低,H2S生成减少;TrxR活性明显降低,心肌组织TRX蛋白表达下调,TXNIP蛋白表达明显上调,介导ROS的产生的NOX-4表达增强,LPO含量明显升高,GSH含量明显降低,细胞凋亡的效应分子和执行者caspase-3表达增强,提示心肌缺血后心肌组织中存在脂质过氧化,并且抗氧化能力减弱,TXNIP/TRX系统参与了心肌缺血损伤的发生和发展;而给予25、50 μg·kg-1DEX干预后,心肌CSE活性升高,内源性H2S生成增多,心肌组织TrxR活性明显升高,TRX蛋白表达上调,TXNIP表达明显受到抑制,NOX-4表达减弱,LPO含量明显减少,GSH含量明显增加,caspase-3表达降低,从而大鼠心脏功能改善,心肌梗死面积减少,心肌组织形态学损伤减轻。与我们的研究结果类似,曹艳丽等[17]报道DEX可以升高血清中H2S水平,对心肺复苏大鼠有一定的保护作用;Yeda等[4]研究证实DEX可以通过调控TXNIP信号减轻肾脏缺血/再灌注损伤;DEX还可以通过抑制TXNIP/NLRP3信号通路对阿霉素所致小鼠心肌细胞损伤具有保护作用[18]。
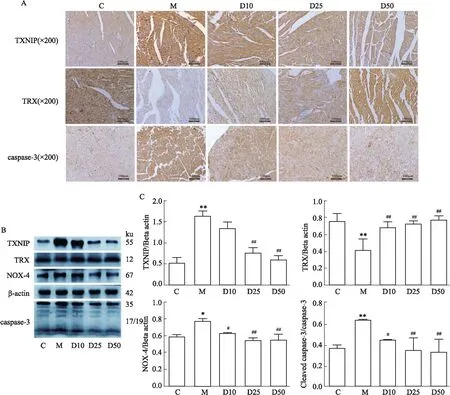
Fig 3 Effects of DEX on TXNIP, TRX, NOX-4 and caspase-3 protein expression in myocardial tissues of myocardial ischemia rats
综上所述,DEX可以促进CSE活性和内源性H2S的生成;促进TrxR活性、增加TRX表达,抑制TXNIP表达,进而催化生成GSH促进谷胱甘肽氧化还原循环,减轻心肌细胞的氧化应激损伤,抑制心肌细胞的凋亡进程,对缺血心肌起到保护作用。但其详细的信号转导过程还有待于进一步采用体外细胞培养、基因敲除技术,通过反向药理学或反向生物学的研究方法证明。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
中国生殖健康(2020年7期)2020-12-10
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国临床医学影像杂志(2019年1期)2019-04-25
中成药(2018年6期)2018-07-11
天然产物研究与开发(2018年1期)2018-02-02
中成药(2017年8期)2017-11-22
中西医结合心脑血管病杂志(2016年20期)2016-03-01
郑州大学学报(医学版)(2015年1期)2015-02-27
