
Ni/CeO2催化剂的金属-载体界面调控及其低温化学链甲烷干重整性能研究
2022-12-14 07:23:40李睿杰章菊萍李孔斋刘慧利
燃料化学学报 2022年11期
李睿杰 ,章菊萍 ,史 健 ,李孔斋,2 ,刘慧利,2,* ,祝 星,2,*
(1. 昆明理工大学 冶金与能源工程学院, 云南 昆明 650093;2. 省部共建复杂有色金属资源清洁利用国家重点实验室, 云南 昆明 650093)
二氧化碳(CO2)和甲烷(CH4)是重要的温室气体和碳源。CO2转化为化学品或中间体需要还原,而CH4的转化则需要氧化。甲烷干重整(Dry Reforming of Methane,DRM)是利用这两种温室气体相互转化的理想方式,反应的产物合成气(Syngas)具有较低的H2/CO比,适合通过费托合成(Fischer-Tropsch synthesis)和羰基合成(Oxo-synthesis)制备液态燃料。然而,甲烷干重整中难以避免副反应发生,在高反应温度下会发生甲烷裂解反应以及一氧化碳歧化反应,这两种副反应都会导致积炭或催化剂失活[1,2]。活性组分在高温下的烧结是催化剂失活的另一个原因[3]。此外,CO2(反应物)和氢气(产物)同时存在引起的逆水煤气变换反应会导致选择性降低,进料气体的转换会受到共进料模式下平衡转换的限制[4]。因此,需要新的工艺路线来提高甲烷和二氧化碳的转化率[5-7]。
在215名参加城乡居民合作医疗保险的高血压患者中,CHE的发生率为13%,其中,农村户口患者CHE发生率为74%; 发生CHE的logistic回归模型,以患者的社会人口经济学特征、疾病严重程度、医疗费用支出等作为解释变量。结果如表4,其中,患者家庭CHE发生的概率随着收入水平的增加而逐渐降低,门诊自付费用每增加一元,其CHE发生的概率将增加0.12%。另外,丧偶状况下的患者发生CHE的概率大于对照组。
首先,消费者应实时了解国家相关政策,以便知晓和维护自己的合法权益。问卷调查结果显示,消费者对政策的变动普遍关注度较低,然而,消费者的这种心理一方面会或多或少的对消费者自身长期利益造成损害,一方面也不利于政策的有效推行和政府的有力管控。
化学链甲烷干重整(CL-DRM))工艺,利用化学链交替进料工艺替代CH4-CO2共进料模式[8-10],具有过程强化、产物分离等优势,克服了传统甲烷干重整共进料模式中的上述缺点[11]。在典型的CL-DRM工艺中,CH4被储氧催化剂的晶格氧选择性氧化并生成合成气,然后失去晶格氧的储氧催化剂被CO2重新氧化[10],完成一个循环。化学链氧化还原循环推动CH4和CO2转化为合成气,H2/CO物质的量比为2[12]。该过程总体上与传统DRM相对应,但实际上由两个独立的气-固反应组成。与传统DRM相比,CL-DRM能在氧化步骤中通过CO2将CH4裂解产生的积炭消除,在一定程度上能减少积炭的产生,而且还能够生产出氢/碳比灵活可调的合成气[13]。化学链技术为CH4和CO2转化提供了一条高效途径,对于碳减排和甲烷资源化将产生重要影响。最近,Buelens等[13,14]提出了一种新型的“超干”甲烷重整工艺,以提高CH4和CO2所生成 CO的产量,该工艺每个CH4能够利用三个CO2分子,并提供了很高的CO空时产率。在这一过程中,Ni基催化剂为甲烷重整催化的活性中心;Fe物种为储氧催化剂,起到氧化还原中心的作用;CaO起到捕获和释放CO2的作用,使氢与碳氧化合物分离,并使得主反应的平衡向正向移动,提高CO2利用率[15]。由于CH4结构稳定,通常化学链干重整工艺需要在600 ℃以上温度下进行,反应温度通常为650-900 ℃,较高的反应温度不仅给催化剂材料带来巨大挑战,同时也降低了工艺能量利用效。
一个典型的涉及载氧剂的化学链过程是由两个或两个以上的还原和氧化步骤组成的氧化还原循环。最简单的形式是,氧载体或氧化还原催化剂首先在低氧分压环境下提供晶格氧生成CO和H2。还原氧载体随后暴露于氧化剂以补充其晶格氧,从而完成两步氧化还原循[11]。在高氧分压情况下,甲烷与氧载体发生完全燃烧反应生成H2O和CO2。从热力学角度出发,氧载体/氧化还原催化剂的供氧能力可通过反应MeOx生成 MeOx-1+1/2O2。高pO2的氧化还原只能用空气再生,适用于化学环空分(CLAS)、化学环氧解偶联(CLOU)或化学链燃烧反应(CLC)。
氧催化剂,通常在化学链工艺中被称作氧载体,是CL-DRM应用中的关键环节[16]。CL-DRM工艺要求储氧催化剂能够在高温下进行稳定的氧化还原循环反应,除储氧催化剂反应性之外,其成本、毒性、热稳定性和耐磨性也是关键的筛选标准[17]。氧化铈具有优越储氧性能[18],具有Ce3+和Ce4+之间快速可逆交换,广泛应用于化学链、催化氧化、电催化等领域。因此,CeO2催化剂在化学链部分氧化领域中得到了广泛应用,研究者开展了大量研究旨在揭示表面结构与催化性能之间的构效关系。研究表明,通过提高活性组分的分散性或金属-载体强相互作用[19,20](Strong Metal-Support Interactions, SMSI),可以有效地提高铈基催化剂的催化性能。Zhu等[21]研究了Ce改性Fe2O3复合氧化物用于化学链重整,发现Fe-Ce复合氧化物在连续的氧化还原循环中表现出良好的性能,可以同时生产纯氢和高质量合成气。他们将这种优异的性能归因于CeO2和Fe2O3物种之间的强相互作用,这种相互作用通过CeFeO3相的形成增强了氧的迁移率并丰富了氧空位。同样,Dou等[22]也揭示了金属-载体相互作用的关键作用,将其应用于化学链重整制合成气工艺中。铈基、镍基[23]催化剂在化学链部分氧化中表现出优越的性能,但在高温条件下遭遇到材料烧结[24]、失活等挑战。因而,低温化学链工艺研究受到广泛关注[25,26],研究者期望通过催化剂结构设计实现CH4和CO2低温化学链转化。
通过暴露晶面调控可以改变氧空位的浓度、活性晶面的作用和金属-载体效应[27]来影响CeO2的催化性能。最近,研究者在可控合成催化剂、设计催化剂表面结构取得重要进展,可通过晶面效应调控催化性能。已有报道合成了许多低维的CeO2纳米材料[28](例如纳米棒、纳米多面体、纳米立方体和纳米管)。通常,不同形状CeO2在晶体结构中暴露出不同的晶面。例如,CeO2纳米棒倾向于优先暴露四个(110)面和两个(100)面,而CeO2纳米八面体暴露八个(111)面[29]。基于密度泛函理论,不同面上氧空位的形成能遵循顺序(110) < (100) <(111),这些晶面的化学活性遵循相反的顺序,表明在CeO2(110)晶面上更容易形成氧空位。到目前为止,有许多关于CeO2在CO氧化[2]、水煤气变换反应和CO还原NO中的晶面效应的报道。研究发现,CeO2用作载体或促进剂,由于高流动氧的存在,Ni/CeO2复合纳米材料可以明显改善Ni活性物种的分散性并抑制积炭形成[30]。然而,Ni/CeO2复合纳米材料作为化学链过程中储氧催化剂的晶面效应仍然是未知的。不同形貌的CeO2载体表现出不同的氧迁移率和与活性金属颗粒的相互作用强度,从而影响沉积炭的气化和镍颗粒的热烧结。为了更好地理解Ni/CeO2储氧催化剂在氧化还原过程中的构效关系,还需要进一步开展相关研究。
对于相邻双侧边盖驱动方腔流动, 在Reynolds数小于1 000的范围内, 还可以找到两对线性不稳定行波模态, 每一对模态在不同Reynolds数下的线性时间增长率曲线在图6(a)和图7(a)中给出.
为揭示Ni/CeO2储氧催化剂在化学链工艺中的晶面效应,通过催化剂结构调控实现低温化学链干重整,本研究提出调控Ni/CeO2储氧催化剂中CeO2暴露晶面并将其应用于化学链干重整工艺。采用水热法、浸渍法制备了一系列具有不同晶面的Ni/CeO2纳米材料(CeO2纳米多面体、纳米棒、纳米八面体和纳米立方体)。在配有在线质谱仪的固定床反应器中考察了Ni/CeO2催化剂的化学链甲烷干重整活性。通过结构和形貌的演化,探讨了CeO2载体的氧迁移能力与催化活性的关系。
1 实验部分
1.1 催化剂的制备
1.1.1 不同形貌载体CeO2制备
通过Rigaku-MiniFlex600型X射线衍射仪研究储氧催化剂反应前后的晶型变化和物相结构分析,利用Jade软件分析数据结果。 X射线衍射仪阳极靶材使用Cu靶,管电压和管电流分别为40 kV和20 mA,以5(°)/min的扫描速率在10°-90°进行扫描,根据Scherrer方程计算平均结晶尺寸(D):DXRD=Kλ/cosβ。其中,K为Scherrer常数,其值为0.89,一般取1;λ为X射线的波长,Cu靶为0.154056 nm;θ为布拉格衍射角,β为实测样品衍射峰半高宽度。
为了进一步验证方法的可靠性,按照实验方法测定Co光谱分析标准样品系列YSS005-1和YSS005-2中Cu、Fe、Ni、Cd、Zn、Mn、Mg、Si、As含量,标准样品系列采用盐酸进行消解,结果见表12。
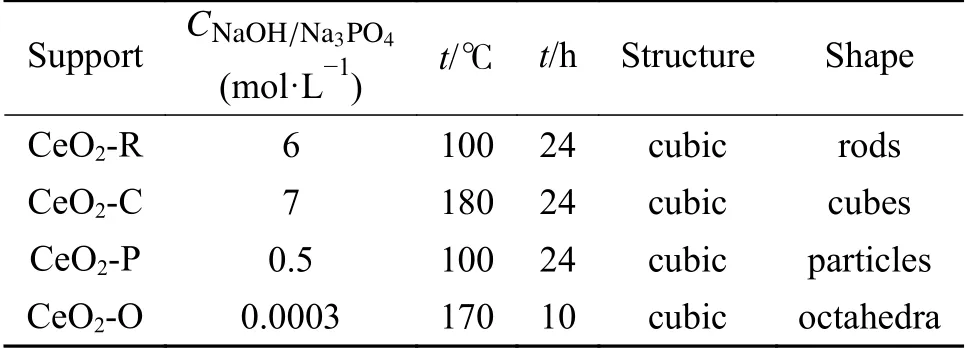
表 1 CeO2纳米结构水热合成条件Table 1 Hydrothermal synthesis conditions of CeO2nanostructures
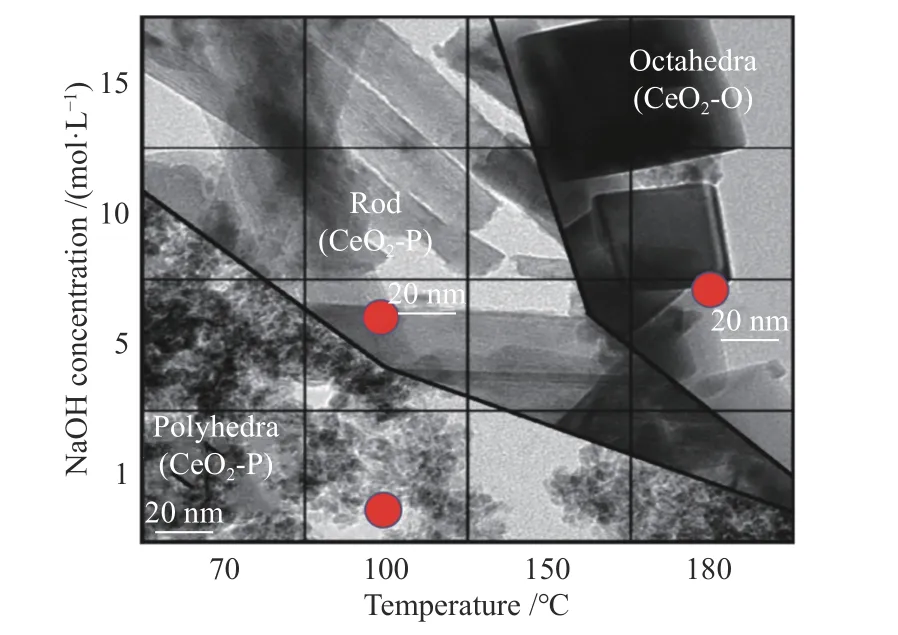
图 1 CeO2水热处理的形貌相图[31]Figure 1 Morphological of CeO2 after hydrothermal treatment[31](with permission from Elsevier)
CeO2纳米八面体制备:CeO2纳米八面体的合成遵循Huang等[32]的方法。将Ce(NO3)3·6H2O (2 mmol)溶于纯水(79 mL)中,然后加入Na3PO4(1 mL, 0.02 m)。将混合溶液在室温下搅拌1 h,然后转移到100 mL聚四氟乙烯瓶中。将聚四氟乙烯瓶紧密密封,在不锈钢高压釜中170 ℃下水热处理10 h。冷却后,收集得到的白色沉淀,用超纯水和乙醇洗涤,在80 ℃真空中干燥16 h。获得的白色粉末在马弗炉中600 ℃下煅烧4 h,合成CeO2纳米八面体。
1.1.2 催化剂的制备
采用浸渍法制备不同纳米结构Ni/CeO2催化剂:将不同纳米结构CeO2粉末与Ni(NO3)2·6H2O溶液在60 ℃磁搅拌下混合,搅拌3 h后,将混合物转移到80 ℃的烘箱中。所得样品在400 ℃煅烧90 min,得到了镍含量为5%的Ni/CeO2纳米材料。将所制得催化剂进行研磨压片,压片成型的压力为10 MPa,破碎,筛分(40-60目)颗粒装入样品袋并贴好标签后续备用。
1.2 催化剂的表征
1.2.1 物相分析
CeO2的制备一般使用可控的水热法合成了不同形貌的纳米CeO2多面体(8个(111)面和6个(110)面)、纳米棒(4个(110)面和2个(100)面)和纳米立方体(6个(110)面),不同水热合成条件见表1。通过在水热处理过程中系统地改变碱(CNaOH)的浓度和温度,随着NaOH浓度和温度的升高,溶解/再结晶动力学的变化,选择性地合成了纳米多面体、纳米棒和纳米立方体,如图1所示。制备所用原料为Ce(NO3)3·6H2O(阿拉丁,分析纯)和NaOH(阿拉丁,分析纯)。将Ce(NO3)3·6H2O溶于纯水,将一定量的NaOH溶于纯水,待NaOH溶液冷却后,将其迅速倒入硝酸铈溶液中,将混合溶液搅拌30 min,形成乳状悬浮液。随后,将混合溶液转移到聚四氟乙烯内衬不锈钢高压釜中,将高压釜转移到温度控制的电炉中,并在100-180 ℃的温度下进行水热处理24 h。
1.2.2 比表面积测定
用SEM和TEM研究了不同CeO2载体的微观形貌,并用HRTEM分析了其结构和暴露晶面。在图4(a)和图4(b)中,观察到CeO2纳米棒的直径为7-19 nm,而长度不均匀为30-120 nm。如图4(c)所示,在CeO2-R上观察到了(200)和(220)面的晶格条纹,它们的晶面间距分别为0.27和0.19 nm。因此,CeO2-R沿(110)方向生长,并主要暴露出稳定性较差的(110)面(位于两个侧面和两个横截面)和(100)面(位于两个侧面)[36],根据CeO2-R的平均尺寸和(110)和(100)面的位置,(110)面和(100)面的比例分别约为54%和46%。 CeO2-C呈现出完美立方体形状结构(图4(d)和4(e))。HRTEM图像显示,晶格条纹对应于(200)和(020)面(属于(100)面),晶面间距为0.27 nm,晶面交角为90°,CeO2-C的形貌为六个(100)面围成的立方体[37]。图4(g)和4(h)显示了尺寸为13-23 nm的均匀CeO2-P的SEM和TEM照片。 HRTEM照片显示,CeO2-P主要暴露出(111)面和(200)面(图4(i))[38]。 图4(l)显示了(111)面晶格条纹,晶面间距为0.31 nm,这表明CeO2-O是由(111)面围成的八面体形状。
1.2.3 形貌分析
使用扫描电子显微镜(SEM)分析、透射电子显微镜(TEM)分析和高分辨率透射电子显微镜分析(HRTEM)观测储氧催化剂颗粒的微观形貌以及暴露的晶面。扫描电子显微镜(SEM)分析在Philip公司生产的NOVA NANOSEM 450进行。透射电子显微镜(TEM)和高分辨率透射电子显微镜(HRTEM)分析在FEI Tecnai G2 F30透射电子显微镜上以300 kV的加速电压进行。
1.2.4 拉曼光谱分析
使用赛默飞世尔科技(中国)有限公司的Thermo Fisher DXRxi型拉曼光谱仪测量材料表面上CeO2载体的性质,所用的激光波长为532 nm,功率3 MW。光谱为100-1600 cm-1,光谱分辨率为5 cm-1,曝光时间为0.1 s。
1.2.5 CH4程序升温还原
为了评价储氧催化剂对甲烷的反应活性,进行了甲烷程序升温还原(CH4-TPR)实验。评价装置主要由固定床反应器、自动进气系统以及检测反应前后气体组成的尾气分析系统三部分组成。测试时,使用200 mg样品,并将气体流量设置为50 mL/min。首先,用Ar冲洗反应器,然后将气体改为5% CH4/Ar。 接着,以10 ℃/min的升温程序从室温升温至800 ℃。使用放置在反应器内的K型热电偶监测样品的实际温度。使用在线质谱仪(LC-D200M PRO, TILON)对出口气体分析进行,该质谱仪采用电子冲击电离、四极杆质量过滤器和二次电子倍增检测器。在CH4-TPR过程中跟踪了以下m/z值:2(、15( C)(m/z15信号优于m/z16信号,以避免与CO、CO2、H2O和O2的交叉影响)、在进行上述反应前,质谱仪的质谱信号用已知组成的标准气体进行标定。通过整合相应的质谱信号,计算出上述反应中的产物(CO、CO2和H2)和未反应的甲烷(CH4)量。
1.3 催化剂的评价
其中,C为气体的实时含量,F为气体的体积流量,R为理想状态下气体的摩尔体积(22.4 L/mol)。
反应中甲烷和二氧化碳的转化率、合成气选择性和收率按照下列公式进行计算:
1.2.1 一般情况调查表 包括年龄、职称、学历、护龄、编制、医院种类、科室、收入、职务、翻班与否和日工作时间等。

化学链甲烷干重整试验与程序升温试验类似,并采用相同的反应器系统。实验使用200 mg样品在1.01325 × 105Pa和550 ℃下进行,试验前用50 mL/min流量的纯Ar吹扫反应器。样品在5% CH4/Ar下等温还原,在5% CO2/Ar下进行等温氧化,气体流量为50 mL/min。还原和氧化周期为10 min,同时在每个半周期之间用惰性气体(Ar)吹扫反应器10 min,以避免还原和氧化气体的混合。出口气体用质谱仪(LC-D200M PRO, TILON)进行分析,质谱仪用标准气体混合物进行系统标定。在安捷伦7890B气相色谱仪中使用气体采样袋定期离线分析气体成分,以检查分析的准确性。
甲烷转化初期会产生一定量的CO2,然后CO和H2同步成为主要产物。一般情况下,CO2的生成来自于甲烷被活性表面吸收氧(式(6))的完全氧化。CO和H2的生成是由于晶格氧(式(7))的作用,可以选择性地将甲烷转化为合成气[46]。在甲烷选择性氧化过程中,CH4转化步骤中的H2/CO物质的量比高于理论值,说明甲烷在此气固反应过程中出现裂解反应(式(8))。甲烷与储氧催化剂之间的气固反应经历了开始的完全氧化和同时的甲烷部分氧化和裂解[47]。在不同的Ni/CeO2样品中,Ni/CeO2-P催化剂与甲烷反应活性最高,产生了最高浓度的CO和H2。在CO2氧化半周期中,还原的储氧催化剂可以被CO2恢复,同时伴随着CO的生成。在还原步骤中沉积在储氧催化剂上的碳也可以通过与CO2反应而被“烧掉”。
2 结果与讨论
2.1 Ni/CeO2储氧催化剂的晶体结构与织构特性
图2为不同形貌的CeO2载体及相应的Ni/CeO2储氧催化剂的XRD谱图。如图所示,在600 ℃下煅烧,通过水合氧化物(CeO2·nH2O)的脱水、无水氧化铈(CeO2)的结晶和Ce3+的氧化、残余硝酸盐分解,充分将水热沉淀物变成晶型CeO2。 新鲜样品中衍射峰都来自于CeO2和NiO相。 CeO2-R、CeO2-C、CeO2-O和CeO2-P在28.7°、33.2°、47.6°、56.5°、59.1°、69.4°、76.6°和79.2°处的衍射峰分别对应于立方CeO2相(JCPDS 34-0394)的(111)、(200)、(220)、(311)、(222)、(400)、(311)和(420)晶面的衍射。按Scherer公式计算的不同样品的晶面尺寸列于表2。四种CeO2载体的衍射峰位置几乎相同,但其相对强度不同。
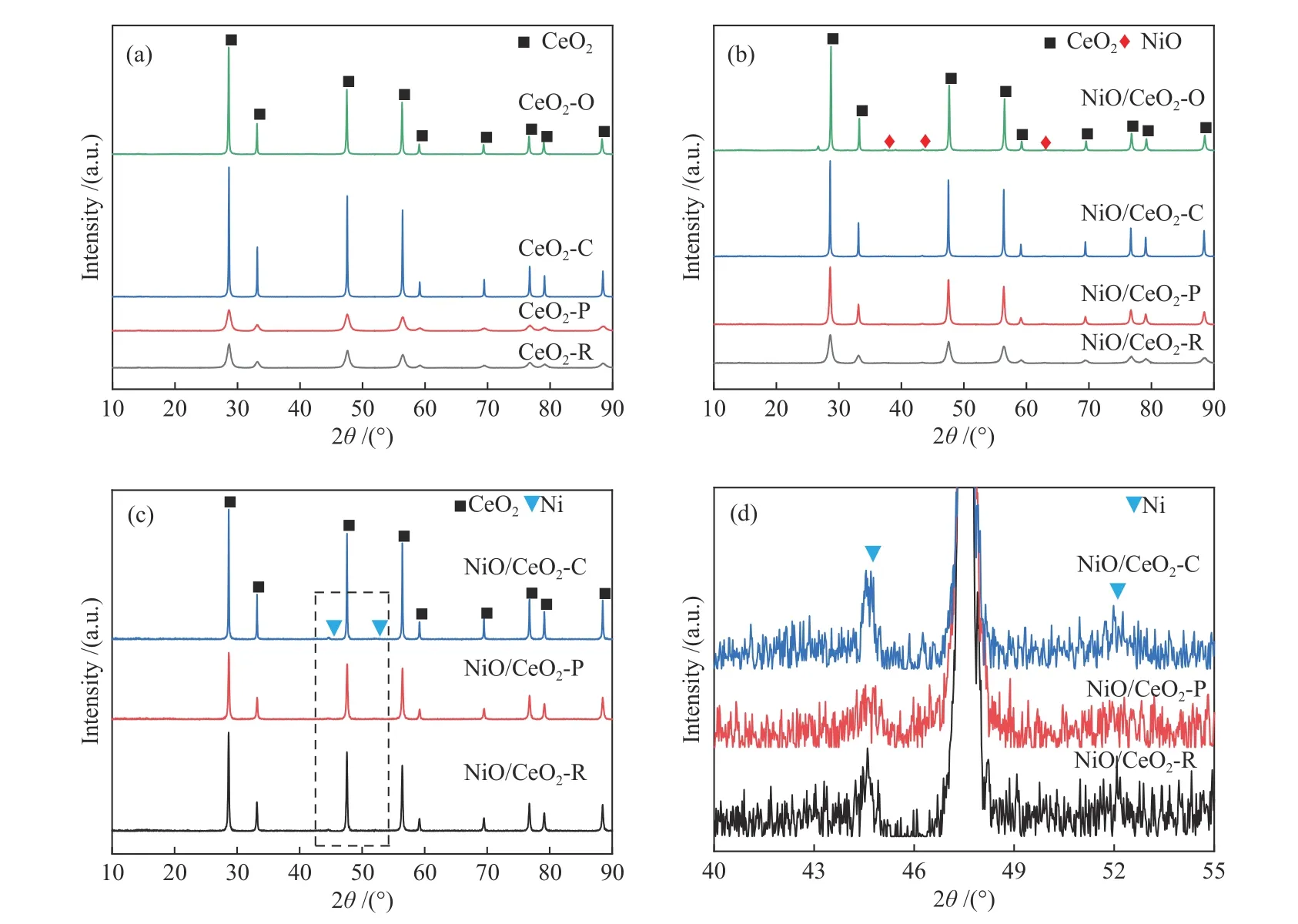
图 2 (a)CeO2、(b)NiO/CeO2和((c)、(d))Ni/CeO2的XRD谱图Figure 2 XRD patterns of (a) CeO2, (b) NiO/CeO2, and ((c), (d)) Ni/CeO2
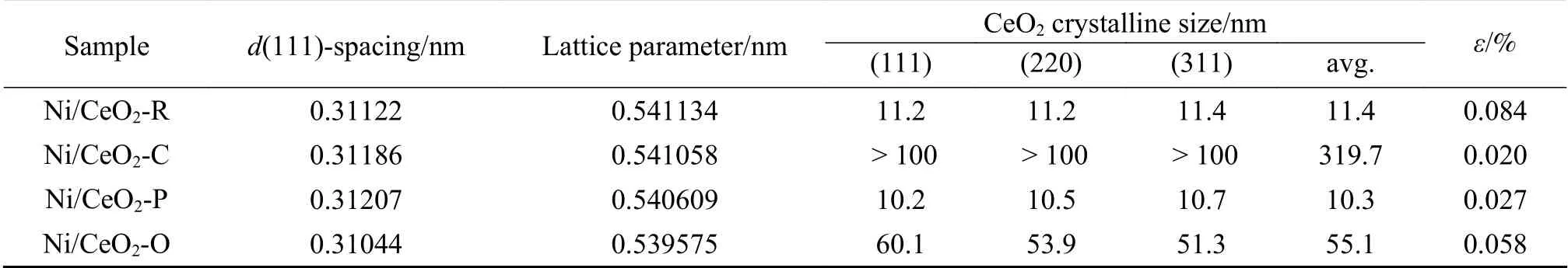
表 2 Ni/CeO2样品中CeO2的晶格参数(a0)、结晶尺寸和微应变(ε)Table 2 Lattice parameter (a0), crystalline size, and the microstrain (ε) of ceria in Ni/CeO2 samples
CeO2-O的(I(200)/I(111)+I(220)/I(111))比值高于CeO2-R、CeO2-C和CeO2-P。说明纳米多面体中的(111)面暴露量高于纳米棒、纳米立方体和纳米多面体,这与图3中TEM、SEM和HRTEM分析结果一致。CeO2纳米立方体表现出最尖锐的峰,强度最高,说明其结晶度最高,晶体尺寸最大。CeO2纳米多面体的衍射峰较宽,意味着与其他三种CeO2纳米材料相比,CeO2纳米多面体的晶体尺寸较小。不同形貌的Ni/CeO2催化剂的XRD谱图如图2(b)-(d)所示,观察到CeO2的衍射峰没有明显的变化,说明CeO2载体的结构没有改变。此外,在Ni负载后,观察到Ni结晶相的一些微弱和宽的衍射峰,可能表明Ni物种在CeO2纳米材料上高度分散。氧化铈的晶格参数(α0)下降(表3),衍射峰的位置略有移动,晶格收缩是由于较小离子半径(rNi2+=0.72)Ni掺杂至氧化铈晶格(rCe4+=0.94)[33]。另外,与纯氧化铈纳米材料相比,Ni/CeO2中铈的结晶尺寸增大,可能与热煅烧过程中的轻微烧结有关。如表2所示,Ni/CeO2-R在四种催化剂中微应变最高,CeO2-C的应变最小。根据之前的报道,应变是测量材料中存在的晶格应力,由于晶格变形、伸长或收缩[34]。因此,应变的变化意味着CeO2-R中氧空位的浓度大于其他材料。
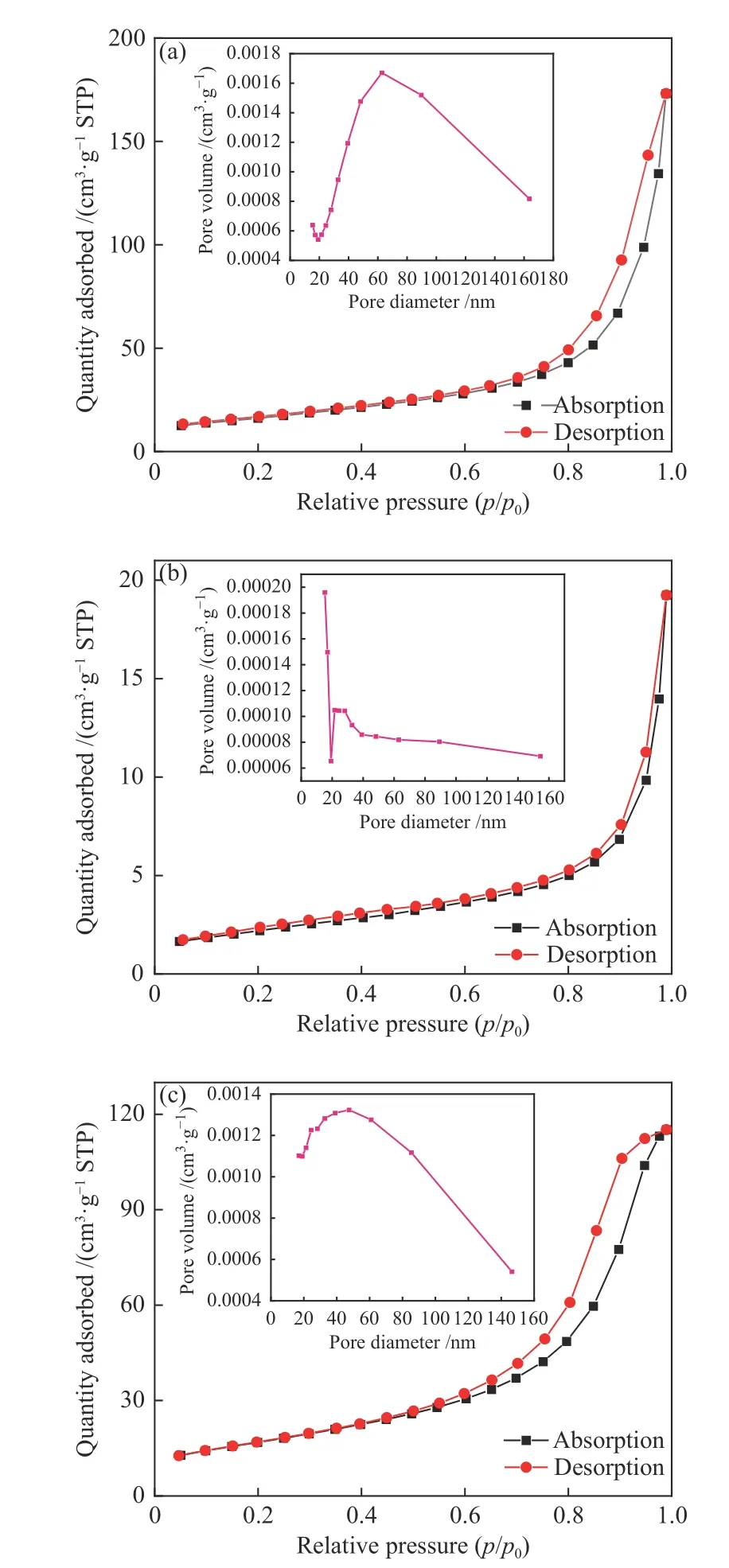
图 3 (a)Ni/CeO2-R、(b)Ni/CeO2-C、(c)Ni/CeO2-P的N2吸附-解吸等温线和孔径分布(插图)Figure 3 N2 adsorption-desorption isotherms and pore size distribution (inset) of (a) Ni/CeO2-R, (b) Ni/CeO2-C,(c) Ni/CeO2-P
表3中提供了包括比表面积(SBET)、孔体积和平均孔径在内的织构特性。一般来说,这些物理特性对催化剂的活性有重要影响。四种催化剂中,Ni/CeO2-P表面有多个缺陷并表现出最高的比表面积,为60.91 m2/g,而Ni/CeO2-C由于颗粒较大,表面相对光滑,表现出最低的比表面积7.93 m2/g(图3)。与Ni/CeO2-P相比,Ni/CeO2-R比表面积达到 57.88 m2/g,粒径达到61.0 nm。大的比表面积有利于NiO物种在载体材料上的分布,有利于其气固催化反应进行。不同Ni/CeO2催化剂的表面积遵循以下顺序:Ni/CeO2-P > Ni/CeO2-R > Ni/CeO2-O > Ni/CeO2-C。图3为煅烧Ni/CeO2催化剂的N2吸附-脱附等温线。所有的样品都表现出IV型等温线,并出现了H1滞后环。IV类型等温线的滞后环表现出介孔结构的特征。在相对压力高于0.65的情况下,通过多层填充滞环,在中间相对压力下吸附了大量的N2。根据IUPAC的H1型滞回环通常与多孔材料有关,这些多孔材料由定义明确的圆柱状孔道或近似均匀的球体团聚而成。插图为相应等温线的BJH孔径分布。这里没有显示纯CeO2的比表面积,但发现在CeO2上负载Ni催化剂的比表面积均有所减小,这是因为负载Ni后,Ni 颗粒阻塞了CeO2载体的孔道所致,Ni /CeO2-P呈现出相对较高的比表面积和孔体积,CeO2-P载体更有利于Ni颗粒分散且不易引起团聚。Ni /CeO2-P 催化剂中含有较多的介孔。据报道,增加介孔数量可提供更多可用的活性位点,从而增加甲烷化反应的活性。Fukuhara等[35]也报道了类似的现象,即在纯CeO2(126.4 m2/g)上分散Ni后,样品的比表面积明显降低(93.4 m2/g)。

表 3 煅烧后的Ni/CeO2催化剂的织构性能Table 3 Textural properties and redox behaviors of calcined Ni/CeO2 catalysts
2.2 Ni/CeO2储氧催化剂的形貌分析
使用美国Quantachrom Instruments公司生产的Quantachrome Autosorb-iQ型物理吸附仪对样品进行N2吸附-脱附测试和BET比表面积测定。样品首先在120 ℃下抽真空干燥3 h,再在300 ℃温度下处理3 h,最后在液氮-196 ℃温度下分别测定氮气吸附分压p/p0在0.1、0.2和0.3时的脱附峰面积并记录校正气体量和校正峰面积然后根据BET公式计算比表面积。
图5中的TEM和HRTEM照片显示,引入镍后,不同形貌的CeO2仍保持了原有的微观形貌。没有观察到镍的聚集现象,说明镍物种的分散度很高。由于CeO2-R表面具有丰富的氧空位,而氧空位可锚定Ni活性颗粒,进而强化了Ni与CeO2-R之间的SMSI效应[39]。

图 4 新鲜制备的CeO2的SEM、TEM和HRTEM照片Figure 4 SEM, TEM and HRTEM images of as-obtained CeO2 ((a), (b), (c)) CeO2-R, ((d), (e), (f)) CeO2-C, ((g), (h), (i)) CeO2-P and((j), (k), (l)) CeO2-O

图 5 Ni/CeO2的TEM和HRTEM照片Figure 5 TEM and HRTEM images of Ni/CeO2((a), (d)) Ni/CeO2-R, ((b), (e)) Ni/CeO2-C and((c), (f)) Ni/CeO2-P
2.3 Ni/CeO2储氧催化剂的氧空位浓度
在Ni/CeO2催化剂中,除了金属活性位点外,氧空位也可能作为一个重要的活性位点。Ni颗粒附近氧化铈的氧空位被认为是干重整的活性位点,当甲烷在催化剂表面被氧化时,氧化剂是催化剂的表面氧原子,从而产生表面氧空位(Mars-van Krevelen机制),气体氧分子通过以下过程供给氧化铈晶格氧空位比正常的氧化物位点更强烈地结合吸附物并有助于它们的解离。采用拉曼光谱来证实Ni/CeO2催化剂的晶面对活性位点密度(氧空位浓度)的影响,如图6所示。
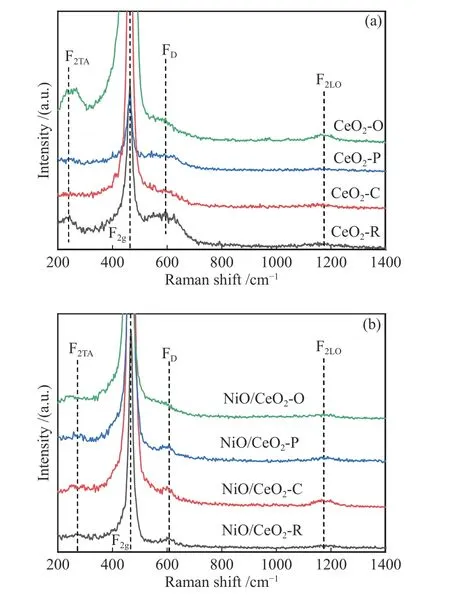
图 6 (a)CeO2样品,(b)Ni/CeO2样品的拉曼光谱谱图Figure 6 Raman spectra of (a) CeO2 samples(b) Ni/CeO2 samples
在CH4-TPR曲线中,储氧催化剂中的非选择性氧化和选择性氧化分别对应于甲烷在初始阶段完全氧化为CO2和H2O,以及在后续阶段选择性氧化为CO和H2,同时,表面金属Ni有利于甲烷重整,也会在一定程度上提高CH4-TPR过程中合成气产量[45]。所有Ni/CeO2样品的反应主要产物为CO和H2,只有在开始反应阶段有少量CO2,这说明Ni/CeO2储氧催化剂对甲烷转化为合成气具有非常高的选择性。同时观察到H2浓度是CO浓度的两倍以上,说明由于甲烷裂解的发生,产生了氢气和积炭。
(2)M=m′∪Part∪Component∪Element,其中:Part表示部件节点的集合,Component表示组件节点的集合,Element表示零件节点的集合。整机由多个部件组成,部件由多个组件和零件组成,组件又由多个零件组成。
2.4 CH4程序升温还原
为研究催化剂的甲烷活化能力,开展了四种催化剂的CH4-TPR实验,如图7所示。不同形貌的Ni/CeO2催化剂上甲烷反应活性随温度而升高,其中纯的CeO2在该反应条件下表现微弱的反应活性,甲烷活化产物(CO和CO2)可忽略不计(未显示)。Ni/CeO2催化剂都呈现出比在纯CeO2样品更高的反应活性,说明Ni有助于活化甲烷从而促进甲烷氧化[44]。从图7(a)-(c)可以看出,当温度低于480 ℃时,四种储氧催化剂几乎无反应活性。随着温度的逐渐升高CO体积分数开始增加,表明Ni负载可促进Ni/CeO2储氧催化剂在低温下释放晶格氧及选择性氧化能力。四种样品的甲烷部分氧化起始温度排序为Ni/CeO2-P(483 ℃)< Ni/CeO2-R(493 ℃)<Ni/CeO2-C(607 ℃)< Ni/CeO2-O(692 ℃)。
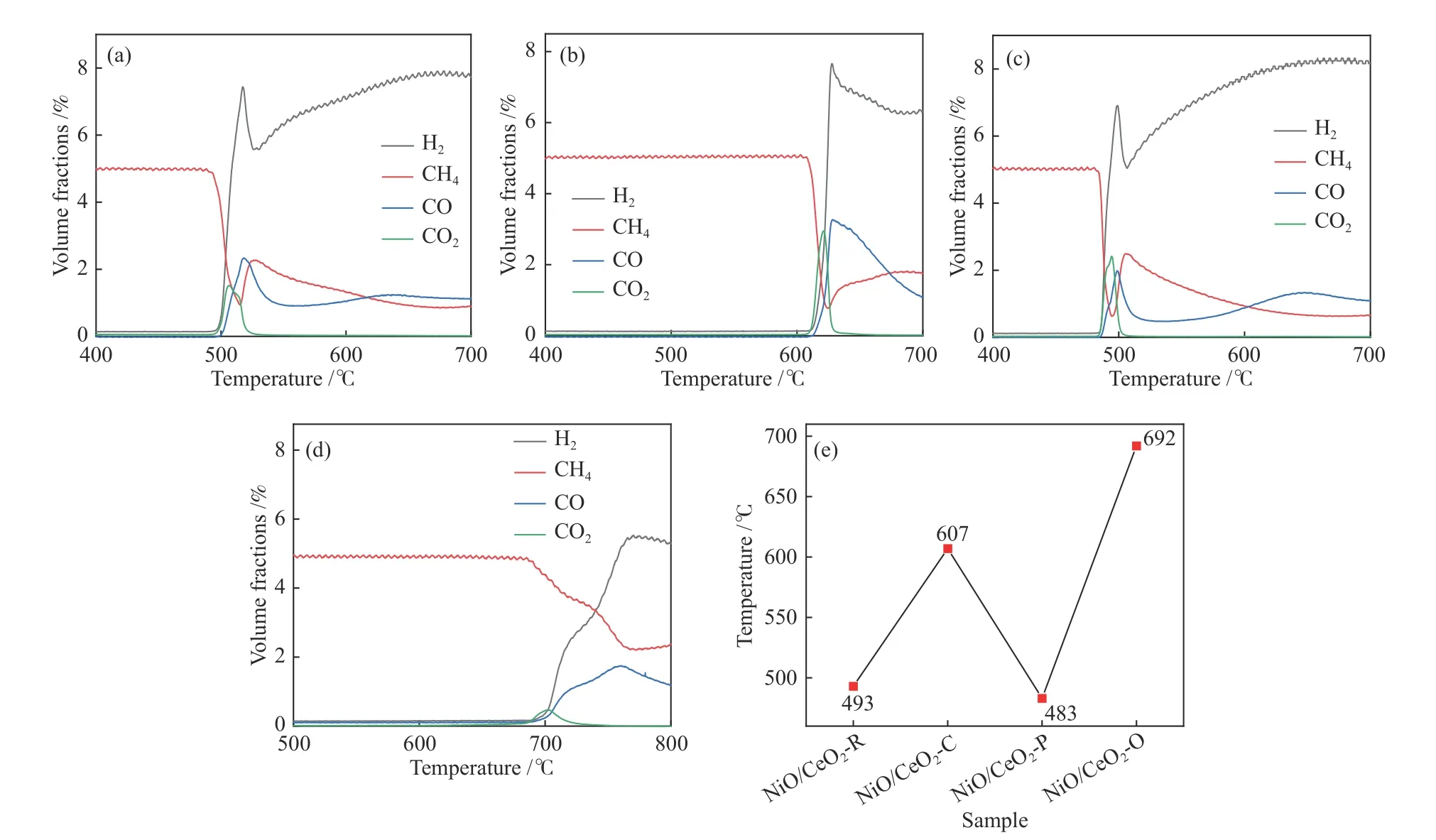
图 7 CH4的温度程序反应(CH4-TPR)曲线:(a)Ni/CeO2-R,(b)Ni/CeO2-C,(c)Ni/CeO2-P;(d)Ni/CeO2:(e)CH4-TPR过程中生成物形成的起始温度Figure 7 Temperature-programmed reaction of CH4 (CH4-TPR) profiles over (a) Ni/CeO2-R, (b) Ni/CeO2-C, (c) Ni/CeO2-P;(d) Ni/CeO2, (e) starting temperature of products formation during the CH4-TPR
对于所有不同形态的CeO2载体,465 cm-1处的强峰与CeO2的一阶F2g对称性有关,位于262、598和1180 cm-1处的三个弱峰分别被分配给CeO2的fcc萤石相的二阶横向声学(2TA)模式、缺陷诱导(D)模式和二阶纵向光学(2LO)模式[41]。FD峰与F2g峰的积分面积之比(标记为ID/IF2g)代表了CeO2表面氧空位的相对浓度,表明CeO2表面存在大量氧空位。与CeO2载体相比,由于Ni种与CeO2之间的强相互作用,Ni/CeO2催化剂的ID/IF2g比值明显增大[42]。此外,ID/IF2g比值受CeO2支撑物的形态影响很大,按以下顺序变化:Ni/CeO2-R > Ni/CeO2-C >Ni/CeO2-P > Ni/CeO2-O。这些结果揭示了不同的CeO2晶面影响了Ni种和CeO2之间的相互作用,导致Ni-CeO2界面周围CeO2表面的氧空位浓度不同。这些结果与DFT计算结果一致,即CeO2表面的氧空位形成能遵循(110) < (100) < (111)的顺序,表面氧空位在CeO2(100)和(110)表面上更易形成,而在CeO2(111)表面上更稳定[43]。在Ni/CeO2催化剂的拉曼光谱中,无法观察到与Ni-O振动有关的特征带。
2.5 化学链甲烷干重整反应
所有Ni/CeO2储氧催化剂在550 ℃下进行CLDRM反应,CH4转化步骤和CO2分解步骤中CH4、H2、CO和CO2气体成分变化情况如图8所示。在CH4转化步骤时,在固定床反应中引入CH4与储氧催化剂反应时,H2和CO的浓度明显增加并达到高浓度,与甲烷的等温反应可大致分为甲烷完全燃烧和甲烷部分氧化两个阶段。甲烷转化步骤中可能发生的反应列成以下公式:
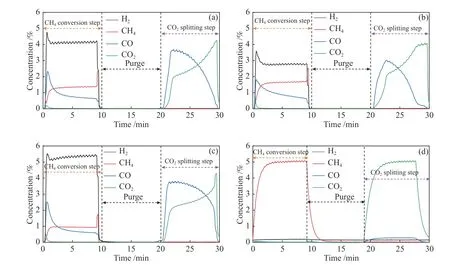
图 8 (a)Ni/CeO2-R、(b)Ni/CeO2-C、(c)Ni/CeO2-P和(d)Ni/CeO2的氧化还原反应产生的气体Figure 8 Gases from the second redox reactions over (a) Ni/CeO2-R, (b) Ni/CeO2-C and (c) Ni/CeO2-P和(d)Ni/CeO2
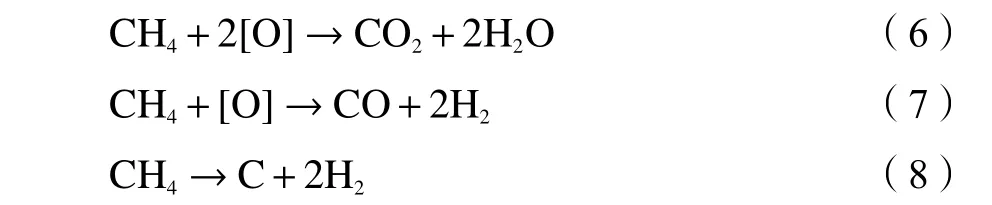
1.2.4 统计学方法 采用SPSS 19.0软件对数据进行分析处理,计量资料以(均数±标准差)表示,采用LSD-t检验;计数资料以(n,%)表示,采用χ2检验,以P<0.05表示差异具有统计学意义。Pearson分析法分析各计量资料之间的相关性。
图9为不同形貌Ni/CeO2储氧催化剂在甲烷氧化(10 min)和二氧化碳分解(10 min)步骤中甲烷转化率、CO选择性和CO2转化率以及合成气产量的定量分析。甲烷转化阶段,晶格氧主要用于选择性氧化甲烷生成CO和H2,少部分活性晶格氧或吸附氧完全氧化甲烷生成CO2和H2O。CeO2具有较强的储氧能力可以与过渡金属金属Ni产生强的相互作用,形成较大的金属支撑面。这些相互作用在Ni原子与氧空位台阶边之间犹为强烈。在铈晶格中,Ni阳离子取代了Ce。将掺杂剂Ni替代到铈晶格中可以激活晶格氧,因为在铈中,大多数Ni-O结合能低于Ce-O结合能[19]。在所有研究中,都采用了传统的掺杂位点结构,其中八面体配位的Ce离子被Ni离子同构取代。除发生氧化反应外,由于晶格氧供给不足或者氧化不及时,部分甲烷裂解产生积炭和H2。CO2分解阶段,CO2恢复CeO2氧空位产生CO,并通过气化反应去除积炭产生CO。由于Ni/CeO2-O在所选择的反应温度下不具备甲烷反应活性,所以相应的结果图中没有提到。对于其余所有的样品,CO的选择性都高于94%。CeO2载体的形态对甲烷转化率影响很大,Ni/CeO2-P的甲烷转化率在三个样品中最高(83.1%)。因此,Ni/CeO2-P样品在甲烷氧化段拥有最高的合成气收率(4.97 mmol/g),在CO2分解段拥有最高的CO收率(2.56 mmol/g)。结果表明,Ni/CeO2-P的催化活性高于其他样品。
(6)学练内容枯燥,缺乏观赏性.前文中提到,白猿通背拳的练习内容主要是由活背八发、四大名山、十字拦、六路总手和十二连炮五种单操组成,现在所习练的白猿通背拳二十四式单操也是由这五种单操为主所创.虽然这些单操也配合着各种对练和打桩进行,去练习白猿通背拳的沉手功夫.但整体来看不难发现白猿通背拳的练习内容并没有清晰的标准,无法用长短高低等外界标准进行衡量且训练形式过于枯燥,导致了其成型套路有了一定的局限性,缺乏观赏性.
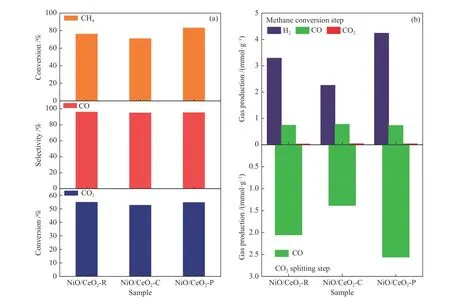
图 9 (a)在不同的Ni/CeO2氧化还原催化剂上,甲烷氧化步骤中CH4的转化率、CO的选择性和CO2的转化率;(b)甲烷氧化步骤中H2、CO和CO2的产率和CO2分裂步骤中CO的产率。Figure 9 (a) CH4 conversion, CO selectivity and CO2 conversion in methane oxidation step over different Ni/CeO2 redox catalyst;(b)Yields of H2, CO and CO2 in methane oxidation step and CO yield in CO2 splitting step
对于Ni/CeO2储氧催化剂而言,在制备条件(煅烧温度、煅烧时间和镍负载量等)相同的前提下,这些催化剂氧化还原反应的活性的差异是源于什么?首先,根据之前的报道,催化活性对催化剂中的金属镍原子数量以及镍颗粒的大小和形状很敏感,但是TEM结果(图5)显示,三种催化剂的Ni分散度几乎相同[48]。从以上结果可以得出结论,本研究中Ni纳米结构不是导致氧化还原反应活性差异的决定性因素。其次,这种差异可能来源于载体CeO2本身的性质差异。首先应考虑载体CeO2的比表面积的影响,所有样品的比表面积为 7.93-60.91 m2/g。根据 Wu 等[49]的文献报道,不同晶面的CeO2的比表面积并不是影响活性和选择性的关键因素,虽然它们的宽度和直径不同,但具有可比性。这是推测比表面积不是影响催化活性的关键参数。根据实验结果和上述分析,这种催化行为差异的主要原因是载体CeO2的晶面效应以及Ni和CeO2之间的相互作用。
载体CeO2的表面结构即形貌(晶面)是影响Ni与CeO2载体相互作用的关键参数。CeO2-R、CeO2-C、CeO2-O和CeO2-P分别暴露出不同的主要晶面,CeO2-R优先暴露四个(110)面和两个(100)面,CeO2-C优先暴露六个(100)面,CeO2-O优先暴露八个(111)面,而CeO2-P主要暴露(111)和部分(100)面。 CeO2的典型结构可视为一组阳离子组成面心立方晶格,氧离子位于四面体间隙位置。对于CeO2的萤石结构,八面体位点是空的并且位于(111)和(110)面或(100)面之间。根据上述结构特点,Ni2+可以很容易地穿透到(110)和(111)面的晶格中,Ni2+的加入是Ni与CeO2之间产生相互作用的重要原因[50]。很明显,Ni在(110)、(100)和(111)面的位点几何和配位环境明显不同。这种结构效应会导致Ni与CeO2之间协同作用的差异,从而影响Ni/CeO2催化剂的催化活性。
2.6 Ni/CeO2储氧催化剂的氧化还原稳定性
由于Ni/CeO2-O不具备反应性,仅对其他三种Ni/CeO2储氧催化剂进行了连续CH4转化/CO2分解氧化还原循环实验,结果如图10所示。与Ni/CeO2-R和Ni/CeO2-C相比,Ni/CeO2-P催化剂具有较高的CH4和CO2的转化率,图12(b)所示,Ni/CeO2-P循环后CeO2主要暴露(111)面,纳米间距为0.31 nm与循环前一致,并没有在氧化-还原出现结构改变,Ni种可以融入到晶格中,Ni/CeO2-P表现出优越的稳定性。其CH4转化率为77.9%-79.1%,经过三次循环后CO2转化率稳定在59.6%左右。Ni/CeO2-R催化剂的CO2和CH4的转化率逐渐降低,Ni/CeO2-R的失活可能是由于Ni颗粒的聚集和积炭的形成,XRD谱显示循环后催化剂表面出现金属Ni衍射峰(图11)。金属Ni颗粒的烧结,导致Ni活性位点数量减少,从而影响了催化稳定性。图10(b)显示,在连续50次氧化还原循环中,两步中Ni/CeO2-P上合成气产量相对恒定,揭示了该储氧催化剂在氧化步骤中再生良好,并在氧化还原循环中保持稳定的结构。
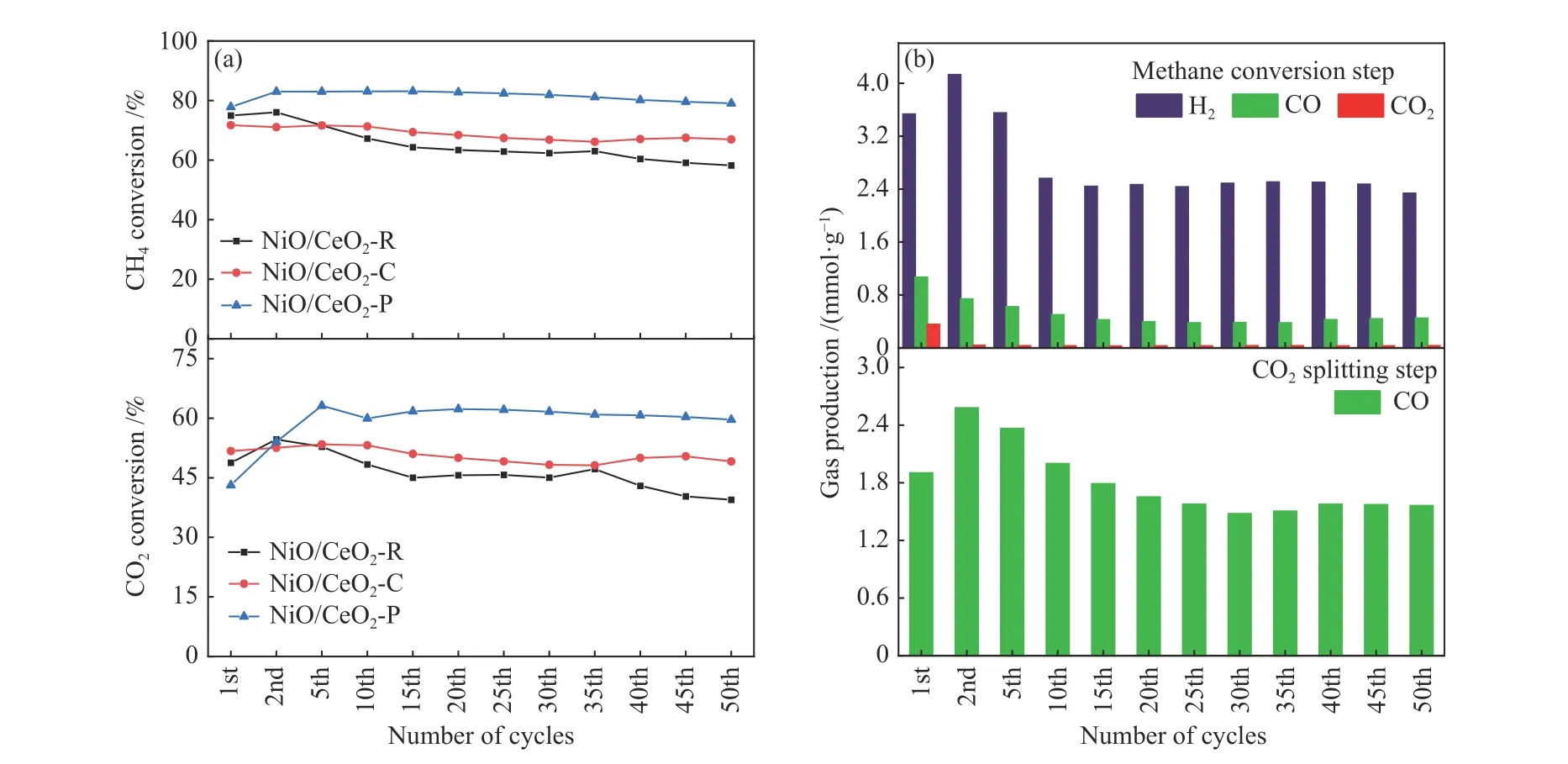
图 10 用于CLDRM的Ni/CeO2氧化还原催化剂的氧化还原稳定性:(a)在550 ℃下,三种Ni/CeO2氧化还原催化剂在连续氧化还原循环中的CH4转化和CO2转化;(b) 在Ni/CeO2-P氧化还原催化剂上,甲烷氧化步骤中H2、CO和CO2的产率以及CO2分裂步骤中CO的产率Figure 10 Redox stability of Ni/CeO2 redox catalysts for CL-DRM (a) CH4 conversion and CO2 conversion in successive redox cycles over three Ni/CeO2 catalyst at 550 ℃; (b) Yield of H2, CO and CO2 in methane oxidation step and CO yield in CO2 splitting step over Ni/CeO2-P catalyst at 550 ℃ for 50 redox cycles
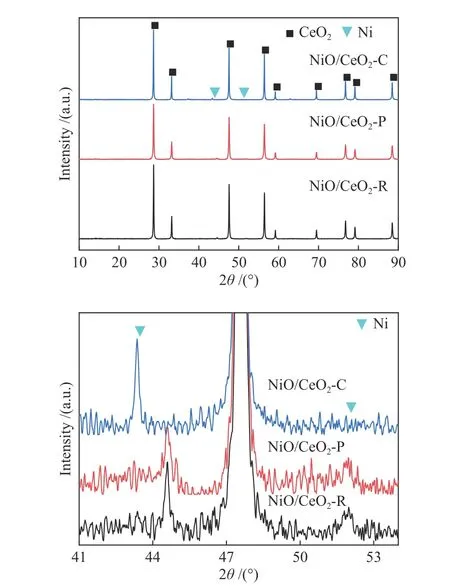
图 11 反应后Ni/CeO2样品的XRD谱图Figure 11 XRD patterns of spent Ni/CeO2 samples

图 12 Ni/CeO2-P的TEM照片Figure 12 TEM images of spent Ni/CeO2-P
3 结 论
本文介绍了一种高活性Ni/CeO2储氧催化剂合成方法,并建立了Ni/CeO2在化学链干重整工艺中的构效关系,揭示了催化剂中CeO2暴露晶面对材料活性及稳定性的影响规律。制备了一系列具有可调载体CeO2形貌/结构Ni/CeO2纳米结构储氧催化剂,并测试了催化剂的化学链干重整性能。
利用水热法、浸渍法制备了不同CeO2形貌(纳米多面体、纳米八面体、纳米棒和纳米立方体)的Ni/CeO2储氧催化剂,结构和形貌表征显示Ni物种高度分散在纳米氧化铈颗粒表面,催化剂比表面积处于7.93-60.91 m2/g范围,其中纳米多面体结构的Ni/CeO2-P比表面积最大。
TRF是肝脏合成的结合金属的糖蛋白。在正常情况下TRF无法通过肾小球滤过膜,因此若在尿液中检测出TRF,则说明肾脏出现一定的损伤。如据钟巧玲[8]试验研究发现,在糖尿病早期肾脏损伤的患者中,检出TRF的数值为(11.38±7.04)mg/L,而在健康人群中,TRF检测数值仅为(0.89±1.22)mg/L。
由于丰富氧空位及Ni-CeO2强相互作用,纳米多面体结构的Ni/CeO2-P储氧催化剂在550 ℃显示出良好的循环稳定性,在CH4转化和CO2分解循环中保持稳定的转化率,材料未出现明显烧结。本研究为高性能Ni/CeO2催化剂设计及其在化学链干重整中应用提供了一条重要途径,特别是低温化学链干重整新工艺对于CH4和CO2转化利用具有积极意义。
猜你喜欢
火炸药学报(2022年5期)2022-11-04 02:30:48
军民两用技术与产品(2021年10期)2021-03-16 06:05:08
水上消防(2020年1期)2020-07-24 09:26:02
数学物理学报(2019年5期)2019-11-29 07:46:50
物理实验(2019年7期)2019-08-06 05:35:56
航空材料学报(2019年2期)2019-04-15 01:04:08
物理学报(2018年22期)2018-12-18 05:58:28
疯狂英语·新读写(2018年3期)2018-11-29 22:37:11
数学物理学报(2017年5期)2017-11-23 07:51:09
潍坊学院学报(2016年6期)2016-04-18 13:56:55
