
N掺杂MoP基核壳纳米棒的制备及其电催化析氢性能
2022-12-14 07:23孙泉锋杨占旭
燃料化学学报 2022年11期
孙泉锋 ,韩 乔 ,杨占旭
(辽宁石油化工大学 石油化工学院, 辽宁 抚顺 113001)
氢气作为可持续再生,无污染的绿色能源载体,有望成为化石燃料的主要替代品[1-3]。电解水制氢作为绿色环保,可再生的技术是实现氢能源的关键[4,5]。近年来,由于贵金属价格昂贵且资源稀缺,难以满足工业化大规模生产需求,过渡金属催化剂逐渐成为研究热点之一,如GO@NiF[6]、MoP/CDs纳米颗粒[7]、Cu/Mo2C/Mo2N多孔结构[8]、MoO3-MoS2纳米球[9]等。
过渡金属磷化物可提供更多不饱和配位原子来驱动电化学析氢反应(HER)[10],其中,最有代表性的磷化钼(MoP),拥有类似Pt的电子结构以及更适中的氢吸附自由能(ΔGH*)去激发析氢活性。例如Huang等[11]通过气相化学沉积法合成的MoP/MoO2/MoS2纳米球催化剂,在10 mA/cm2电流密度下表现出135 mV的过电位。但MoP在高温下易烧结团聚导致活性面积大量损失[12],同时复合物中的惰性组分和材料的弱导电性也会对析氢产生负面影响。为此,研究者们对MoP的表面形态、电子结构等方面做了诸多改进。常用的方法是导电基体复合,如多壁碳纳米管上负载的MoP纳米颗粒[13],分散在多孔炭中的MoP纳米颗粒[14],能够有效降低MoP的烧结团聚并提升电荷传输能力,但这类材料的催化活性依赖于基体调节,炭基体与纳米颗粒之间不稳定的界面阻碍对活性相的进一步调控[15]。另外,杂原子掺杂作为一类直接改变活性相电子结构的方法,通过改变Mo原子d轨道能级来实现ΔGH*可调节[15],例如S、N共掺杂的MoP纳米晶[16],N掺杂MoP纳米颗粒[17],然而高阻抗造成的低效率电荷传输问题却无法解决。这些对MoP单方面改性的材料所展示的性能显然不足以满足目前析氢需求,开展MoP材料的双重调控策略来弥补单方面改性的不足是必要的。最近,Pi等[18]合成的核壳MoP基析氢催化剂,由于其炭层的包覆和界面作用而优化了析氢能力。基于此项研究灵感,利用高氮乙二胺作为中间体合成了N掺杂的MoP基核壳纳米棒催化剂(N-MoP/NC-8)。
具体而言,N-MoP/NC-8纳米棒是通过气-固反应磷化三氧化钼-乙二胺有机无机杂化纳米棒(MoO3/EDA)所制备,乙二胺在高温下形成炭包覆层,炭层可以提高材料电荷转移能力,并将活性相MoP封装,在限域空间效应下实现了强导电客体与活性相之间的原位自然生长以达到两相复合,同时引入了电负性N原子来调节活性相电子结构,完成双重调控以全面改善析氢活性。之后对材料进行了物相、形貌和结构等表征和电化学测试,研究了双重调控对析氢性能的提升作用。
1 实验部分
1.1 实验试剂
三氧化钼(MoO3,AR),乙二胺(EDA,AR),无水乙醇(C2H5OH,AR),次磷酸钠(NaH2PO2,AR),Nafion溶液(C10H8O,AR),氢氮混合气(5% H2+95% N2,高纯),去离子水(自制)。所有药品均自国药集团化学试剂有限公司和阿拉丁化学试剂有限公司购买,气体在本市气体供应站购买,均无需进一步提纯即可使用。
1.2 材料的制备
MoO3/EDA有机无机杂化材料的合成:将12 mL乙二胺溶于150 mL无水乙醇中,搅拌30 min待其混合,加入0.75 g MoO3,在70 ℃下搅拌24 h。反应结束后,趁热抽滤并洗涤,于45 ℃真空干燥12 h,得到MoO3/EDA前驱体。
N-MoP/NC-8复合材料的合成:称取上述0.2 g MoO3/EDA和1.6 g NaH2PO2分别放于管式炉加热区中心的两个瓷舟中,在氢氮混合气氛围下升温至800 ℃,保温2 h,得到N-MoP/NC-8复合材料,制备过程见图1。为了探究材料对析氢反应的最佳合成条件,首先将质量比改变为1∶7和1∶9,再将温度改变为750和850 ℃,分别命名为N-MoP/NC-7、N-MoP/NC-9、N-MoP/NC-750和N-MoP/NC-850。
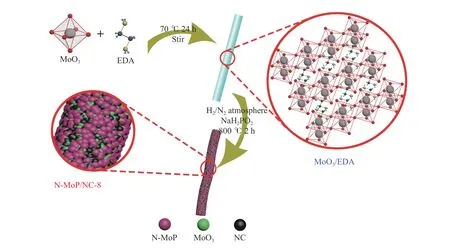
图 1 N-MoP/NC-8的合成示意图Figure 1 Synthesis process of N-MoP/NC-8
MoP-8复合材料的合成:与N-MoP/NC-8合成方案一致,未加入乙二胺。
1.3 材料的表征
Bruker D8型X射线衍射仪(XRD,德国布鲁克公司),采用Cu-Kα1靶,管电压为40 kV,管电流为40 mA,波长λ= 0.154 nm,扫描角度2θ为10°-70°;SU 8010型扫描电子显微镜(SEM,日本日立公司);JEM-2100型透射电子显微镜(TEM,日本电子株式会社);ESCALAB 250型X射线光电子能谱仪(XPS,美国赛默飞世尔科技公司),采用X射线源Al-Kα;DXR型拉曼光谱仪(Raman,美国赛默飞世尔科技公司),激光光源为532 nm,激光功率为9 mW;NOVA4200型表面积及孔隙度分析仪(BET测试)。
1.4 电化学测试
电化学测试在三电极系统的VSP-300型电化学工作站(法国Bio-logic公司)进行。对电极为石墨电极,饱和甘汞电极(SCE)为参比电极,负载催化剂的碳纸(1 cm ×1 cm)为工作电极。工作电极的制备:将5 mg催化剂加入0.5 mL乙醇/Nafion混合液中(体积比9∶1),超声30 min以上使其完全分散,再移取一定量的溶液滴加在碳纸上,利用红外灯干燥后使用,负载量为1.5 mg/cm2。电化学测试均在0.5 mol/L H2SO4溶液中进行,为了保证测试的稳定性,先对材料进行了5 min以上的循环伏安测试(CV)。线性扫描伏安法(LSV)的扫描速率为2 mV/s。电化学阻抗测试(EIS)的频率为100 kHz-100 mHz。稳定性CV循环的扫描速率为200 mV/s。计时稳定电压测试(CP)的电流密度为10 mA/cm2。所有电位均根据公式E(vs. RHE) =E(vs. Hg/HgCl2)+0.244+0.059pH进行标准氢电极电位校正。
2 结果与讨论
2.1 材料结构和形貌表征
图2(a)、(b)为MoO3和前驱体MoO3/EDA的XRD谱图。可以看到,MoO3和乙二胺经过复合后,在11.88°和16.12°处的强衍射峰可对应于乙二胺的有机无机杂化体系[19,20]。这与文献报道的GeOx/EDA材料存在类似结构,即无机金属氧化物骨架与乙二胺中性分子通过氢键复合的杂化结构[21]。为了验证MoO3/EDA的结构,对其进行红外和XPS测试。图S1(c)的FT-IR光谱展示了MoO3/EDA复合材料基团的特征吸收,其中,乙二胺(图S1(a))和MoO3(图S1(b))的特征吸收峰于前驱体MoO3/EDA复合后均发生了偏移,证明MoO3/EDA复合材料中的两种组分之间存在相互作用,在3443 cm-1处O-H伸缩振动的宽高斯型峰说明材料分子间存在氢键[21]。相应的,在MoO3/EDA的Mo 3d(图S2(a))、N 1s(图S2(b))XPS光谱中,232.4、235.5和398.4 eV的峰信号归属于Mo6+的Mo 3d5/2、Mo 3d3/2和Mo 3p轨道[22,23],而399.4 eV为典型的伯胺N结合能,并非烷基铵(- N,402.4 eV)[24],证实乙二胺是以伯胺形式与MoO3通过微弱的氢键复合,这种作用使复合材料的晶体结构发生了改变,形成了有机无机杂化材料。
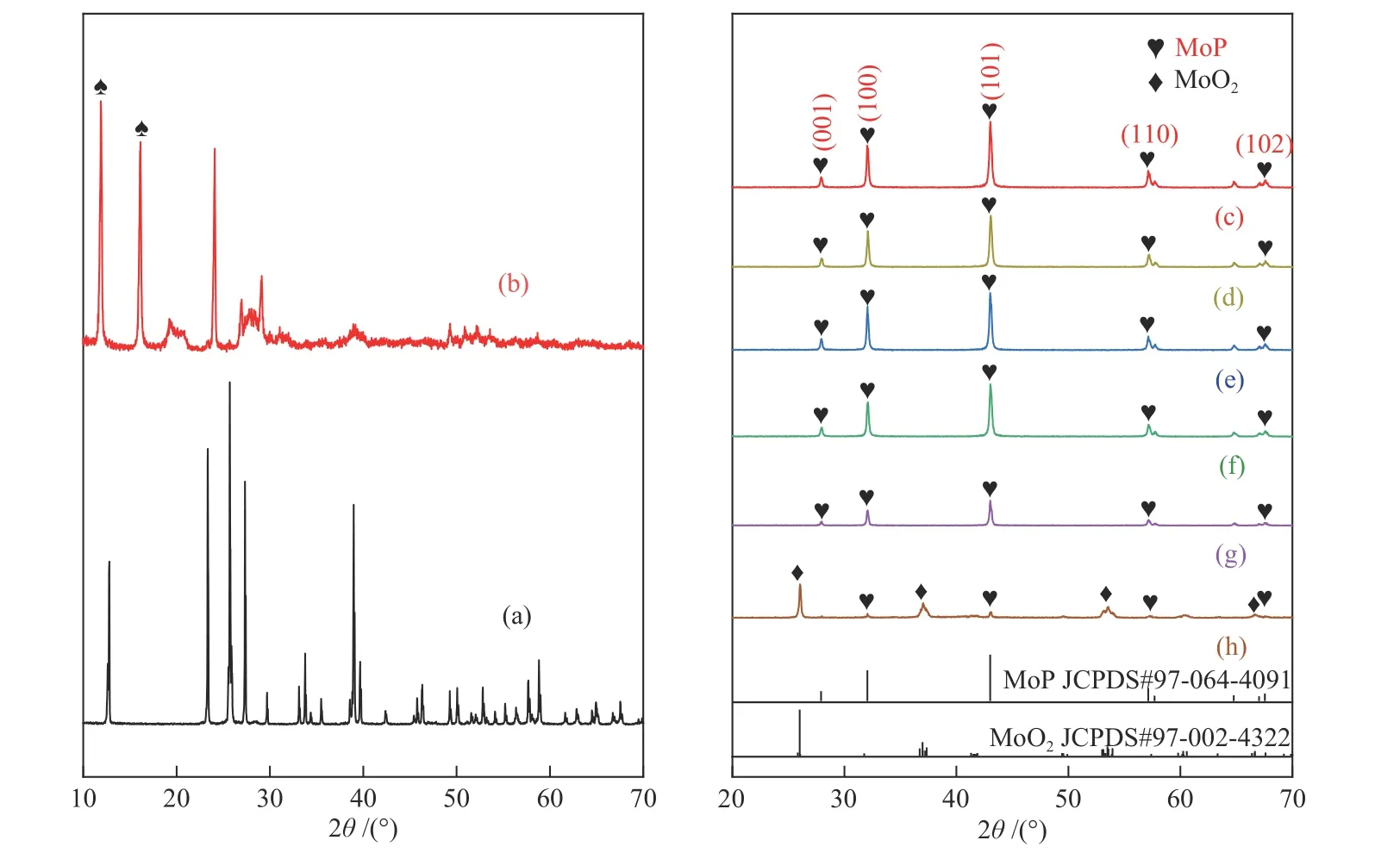
图 2 (a) MoO3,(b) MoO3/EDA,(c) N-MoP/NC-8,(d) N-MoP/NC-7,(e) N-MoP/NC-9,(f) N-MoP/NC-750,(g) N-MoP/NC-850和 (h) MoP-8的XRD谱图Figure 2 XRD patterns of (a) MoO3, (b) MoO3/EDA, (c) N-MoP/NC-8, (d) N-MoP/NC-7, (e) N-MoP/NC-9,(f) N-MoP/NC-750, (g) N-MoP/NC-850 and (h) MoP-8
图2(c)-(g)展示了不同合成条件下对前驱体MoO3/EDA磷化后的XRD谱图,在2θ= 27.9°、32.1°、43.0°、57.1°、67.5°等峰位出现明显的强衍射峰,分别对应六方晶系MoP的(001)、(100)、(010)、(110)、(102)等晶面,与标准卡片(JCPDS NO. 97-064-4091)一致,表明最终产物均为MoP。但不同的是,MoP-8材料由部分MoP以及MoO2物相组成(图2(h)),原因在于高温下磷化氢(PH3)气体对MoO3产生的还原作用[25]。
图3(a)-(h)为MoO3、MoO3/EDA和不同合成条件下MoP的SEM照片。
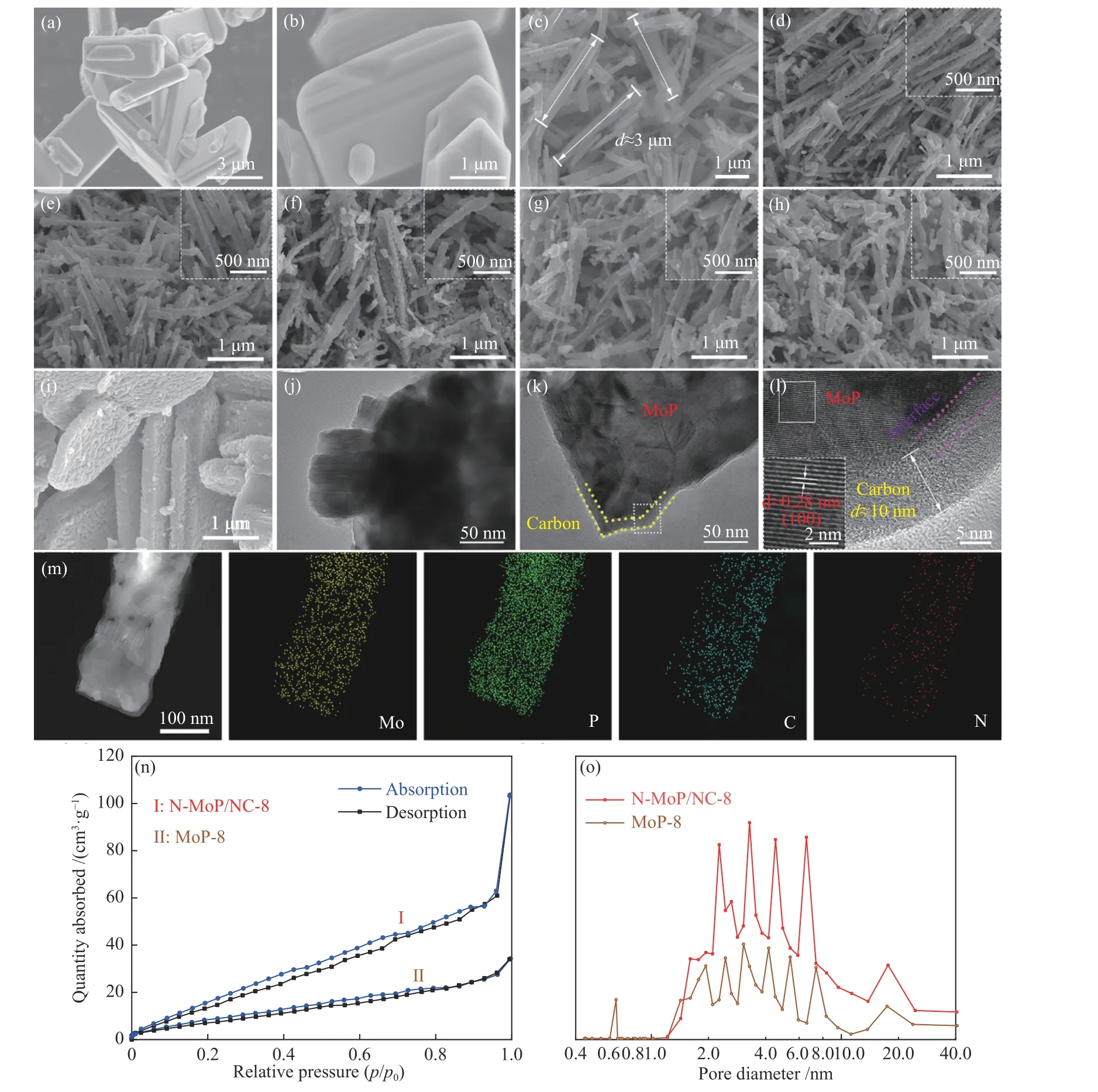
图 3 ((a)、(b)) MoO3,(c) MoO3/EDA,(d) N-MoP/NC-7,(e) N-MoP/NC-8,(f) N-MoP/NC-9,(g) N-MoP/NC-750,(h) NMoP/NC-850和 (i) MoP-8的SEM照片;(j) MoP -8和 (k) N-MoP/NC-8的TEM照片;(l) N-MoP/NC-8的高分辨TEM照片;(m) N-MoP/NC-8的元素EDX mapping分布;N-MoP/NC-8和MoP-8的 (n) N2吸附-脱附曲线和 (o) 孔径分布Figure 3 SEM images of ((a), (b)) MoO3, (c) MoO3/EDA, (d) N-MoP/NC-7, (e) N-MoP/NC-8, (f) N-MoP/NC-9, (g) N-MoP/NC-750,(h) N-MoP/NC-850 and (i) MoP-8; TEM images of (j) N-MoP/NC-8 and (k) MoP-8; HRTEM image of N-MoP/NC-8 (l); Elements EDX mapping of N-MoP/NC-8 (m); N2 adsorption desorption plots (n) and pore size distribution (o) of N-MoP/NC-8 and MoP-8
由图3可见,MoO3表现为类片状的块体形貌(图3(a)、(b)),复合后的前驱体MoO3/EDA呈现为长约3 μm,宽100-200 nm的光滑纳米棒形态(图3(c)),再经不同合成条件的高温磷化,最终MoP材料均保持了纳米棒形貌(图3(d)-(h)),但表面出现锯齿形痕迹,归因于NaH2PO2高温分解的磷化氢(PH3)气体对纳米棒产生的磷化作用以及高温导致的断裂变薄现象。相比之下,材料形貌随合成条件变化显著,出现了低温(750 ℃)下的不充分磷化(图3(g))和高温(850 ℃)导致的磷化过度以及形貌破坏现象(图3(h)),改变原料配比也表现出类似的结果(图3(d)-(f))。图3(k)的TEM照片所示,N-MoP/NC-8呈现核壳结构,外部是无定形包覆层,内部为分散的不规则晶体,其局部高分辨TEM(HRTEM)(图3(l))所示,N-MoP/NC-8内核的晶格间距d=0.28 nm,对应MoP的(100)晶面[26,27],无定形包覆层的平均宽径低于10 nm,且与内核之间存在明显的界面。图3(m)所示为N-MoP/NC-8的一系列元素mapping分布,表明材料含有Mo、P、C、N元素且分布均匀。以上结果证实,N-MoP/NC-8是由无定形炭包覆的MoP晶组成。不同的是,未引入乙二胺的MoP-8材料呈现较厚的带有锯齿形的片状形貌(图3(i)),这与前体MoO3有关,由于气-固反应是从外表面开始进行,这种块状结构会隐藏磷化反应位点,导致材料无法完全转化为MoP物相,对应在MoP-8的TEM图(图3(j))中也并未发现无定形包覆层,内部团聚也较为严重。对比可见,N-MoP/NC-8的炭外壳可以防止MoP高温烧结,减弱堆叠程度。BET测试(图3(n)、(o)和表1)同样证实了此结果,孔径分布(图3(o))表明,NMoP/NC-8和MoP-8基本由小尺寸介孔构成,而纳米棒形貌的N-MoP/NC-8存在更大的比表面积(117.30 m2/g)和孔体积(0.186 cm3/g),这会增加吸附氢中间体(Hads)可能的接触率,促进H2分子的形成、转移和释放,为析氢反应提供场所。
为了对比确定炭层组成,图4是对N-MoP/NC-8和MoP-8进行的Raman光谱测试。N-MoP/NC-8的谱线(图4(a))在拟合后出现D峰(1358 cm-1)和G峰(1584 cm-1),分别对应无序炭和石墨化炭,峰强比(ID/IG≈0.9)反映出炭层的石墨化程度略高于无序化程度(表2)。值得注意的是,在1250 cm-1处出现的D4峰属于D波段的宽低频肩,猜测是N的嵌入,在1500 cm-1处的D3峰存在于高度缺陷的炭中,说明材料存在无定形炭[28]。而MoP-8材料未引入碳元素,因此,其Raman光谱(图4(b))在碳区域不存在振动特征。由此可见,乙二胺炭化形成的炭包覆层可能对材料导电性以及特殊活性位点的激活均存在一定贡献。

表 1 N-MoP/NC-8和MoP-8的比表面积、孔体积和平均孔径Table 1 Surface area, pore volume and average pore diameter of N-MoP/NC-8 and MoP-8
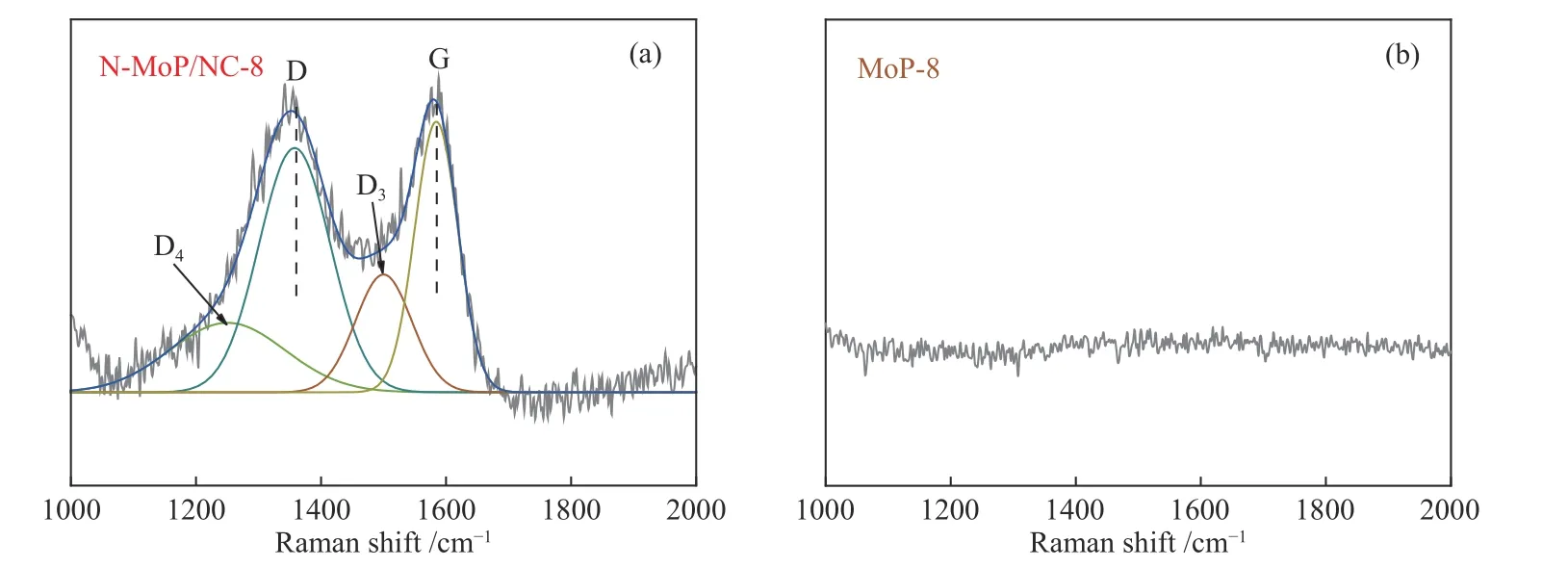
图 4 (a) N-MoP/NC-8和 (b) MoP-8的拉曼光谱谱图Figure 4 Raman spectra of (a) N-MoP/NC-8 and (b) MoP-8
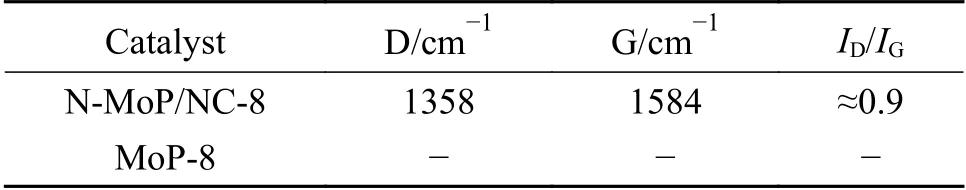
表 2 N-MoP/NC-8和MoP-8的D峰和G峰Table 2 D-peak and G-peak of N-MoP/NC-8 and MoP-8
XPS光谱用于分析N-MoP/NC-8和MoP-8的组成结构和键合情况。如图5(a)所示,在N-MoP/NC-8的Mo 3d谱中,228.6和231.7 eV处的峰对应MoP的Moδ+3d5/2和Moδ+3d3/2轨道[29],236.1和233.5 eV的峰属于MoO3的Mo6+3d3/2和Mo6+3d5/2轨道[16],232.6和229.2 eV的峰对应MoO2的Mo4+3d3/2和Mo4+3d5/2轨道[30]。由于XRD没有测出氧化态物相,微弱氧化态在酸性溶液中应不足以对析氢反应产生影响[31]。在N-MoP/NC-8的P 2p谱中(图5(b)),129.8和130.6 eV处的峰对应MoP的Pδ-2p3/2和Pδ-2p1/2轨道[29],134.2 eV处的峰是由于空气接触而必然产生的P-O键[17,26],在132.9和135.0 eV处分别为P-C键和P-N/PON键,而P的相对原子含量(15.18%)要高于Mo(10.49%)(表3),表明MoP晶格中的P原子和带有电负性的O、C、N等原子成键使结构稳定[16,31],这种强作用力使炭层和MoP相稳定复合。结合材料的Mo 3d、P 2p谱(图5(a)、(b))以及表3给出的结合能数据对比分析,N-MoP/NC-8中MoP的Moδ+、Pδ-轨道结合能与MoP-8相比均存在正偏移,归因于N-MoP/NC-8可能存在电负性更高的原子掺杂,导致其电子密度被撤出,同时N 1s谱(图5(d))的394.5 eV宽峰对应的N-Mo键也说明了有N原子掺入MoP晶格[32]。根据前人有关N掺杂MoP的DFT报告称,这种掺杂会使Mo、P周围的电子云密度向N原子附近聚集,导致Mo的d带中心下降[33],在这种强电子作用的影响下对材料的ΔGH*进行调节,更有利于析氢反应[33,34]。另外,在C 1s谱(图4(c))的C-N/C=N键和N 1s谱(图4(d))中的吡啶N、吡咯N和石墨N来自于掺杂在炭中的N原子[35-38]。据近期的DFT研究表明,在这种类似于炭外壳包覆的MoP核壳结构中,有一些在炭分子层特殊位置的N原子于亚级Mo层调节下拥有近似Pt的ΔGH*[39],因此,N-MoP/NC-8中较高含量的吡啶N(5.41%)作为理论最优N组分可以为HER提供有效的活性位点。XPS的结果与Raman和TEM很好地吻合,证明乙二胺的原位炭化不仅为催化剂提供了限制MoP生长的炭层,同时也是提供N原子的掺杂剂,是双重调节的核心。
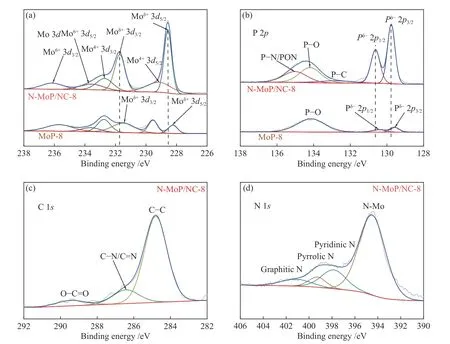
图 5 N-MoP/NC-8的 (a) Mo、(b) P、(c) C、(d) N元素和MoP-8的 (a) Mo、(b) P元素的XPS光谱谱图Figure 5 Mo (a), P (b), C (c) and N (d) XPS spectra of N-MoP/NC-8 and MoP-8

表 3 N-MoP/NC-8和MoP-8的Mo、P组分含量以及结合能差值的XPS光谱数据Table 3 Data derived from XPS spectra of N-MoP/NC-8 and MoP-8
2.2 电化学测试和表征分析
电化学测试前对电解液进行了氮气处理以去除电解液和电极表面的气泡干扰。为了探究材料在析氢电压(0-(-1) V)下的电化学性能,测试了商业的Pt/C催化剂作对比。
为了确定材料对于析氢反应的最佳原料配比,维持800 ℃焙烧温度,分别对原料比1∶7、1∶8和1∶9的材料进行性能测试。过电位(η10)是初步评估催化剂性能的重要参数,LSV极化曲线所示(图6(a)和表4),在电流密度为10 mA/cm2时,Pt/C的过电位为31 mV,这与文献报道的性能接近[40,41]。此外,N-MoP/NC-7、N-MoP/NC-8和N-MoP/NC-9的过电位分别是112、92和111 mV,因此,N-MoP/NC-8的析氢活性最好。Tafel斜率代表电流强度和超电势在低过电势极化区域的线性关系,反映HER动力学快慢[20,42]。由Tafel斜率曲线所示(图6(b)和表4),N-MoP/NC-8的Tafel斜率最低(68 mV/dec),表明N-MoP/NC-8拥有最快的析氢反应动力学。电化学阻抗测试(EIS)反映催化剂的HER电荷转移能力,图6(c)右上为等效模拟电路图,包括表面溶液电阻(Rs)、电荷转移电阻(Rct)以及CPE恒相原件。由图6(c)和表4数据所示,N-MoP/NC-7、N-MoP/NC-8和N-MoP/NC-9的阻抗分别是2.4、2.0和2.6 Ω,其中,N-MoP/NC-8的阻抗值最小,具备最优的电荷转移能力。由此可见,原料比1∶8为材料的最佳合成条件。
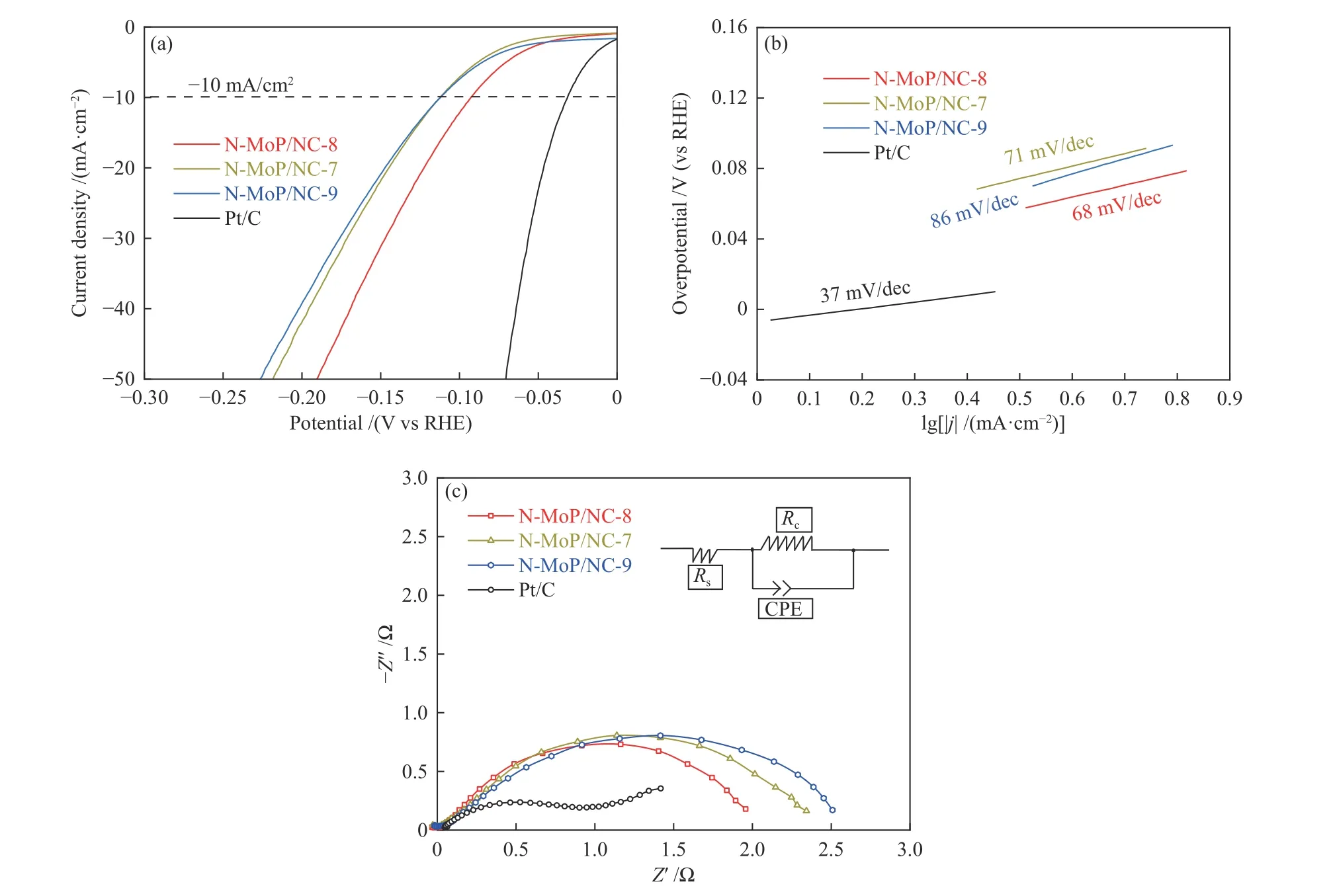
图 6 不同原料比的MoP和Pt/C的 (a) LSV极化曲线,(b) Tafel斜率曲线,(c) EIS曲线Figure 6 LSV polarization curves (a), Tafel slope curves (b), and EIS curves (c) of MoP materials synthesized with different raw material proportions
接下来考察焙烧温度对材料析氢性能的影响,采用1∶8的原料配比,对焙烧温度750、800和850 ℃的材料进行电化学测试,并额外测试了不加乙二胺的MoP-8作为对比来分析材料的双重调控作用。首先,LSV极化曲线(图7(a))对比表明,800 ℃合成的N-MoP/NC-8在所有材料中表现出最好的析氢活性,其过电位为92 mV(表4)。而经过双重调控的样品性能均优于MoP-8(226 mV),这表明在限域空间效应下被炭包覆的MoP,其堆叠程度大幅降低,暴露出更多的活性位点并增大了比表面积和孔体积,提升了析氢性能。其次,酸性电解液的HER包括三个可能的反应步骤:H++e-→Hads(Volmer反 应,120 mV/dec);Hads+H++e-→H2(Heyrovsky反应,40 mV/dec);Hads+Hads→H2(Tafel反应,30 mV/dec)。因此,由图7(b)的Tafel斜率曲线和表4数据所示,MoP材料均遵循典型的Volmer-Heyrovsky机制,HER过程受到解吸反应限制,而N-MoP/NC-8经过N掺杂的电子调节,优化了Hads的转化速率,表现出最低的Tafel斜率(68 mV/dec),未经改性的MoP-8其析氢动力学最差(115 mV/dec)。最后,由图7(c)的EIS曲线所示,N-MoP/NC-8的阻抗谱线半径在所有MoP材料中最小,阻抗为2 Ω(表4),表明其具备更好的电荷转移能力,而MoP-8表现出相对较高的阻抗值,电荷转移能力最差,说明石墨化的碳层促进了材料在析氢过程中的电荷转移。
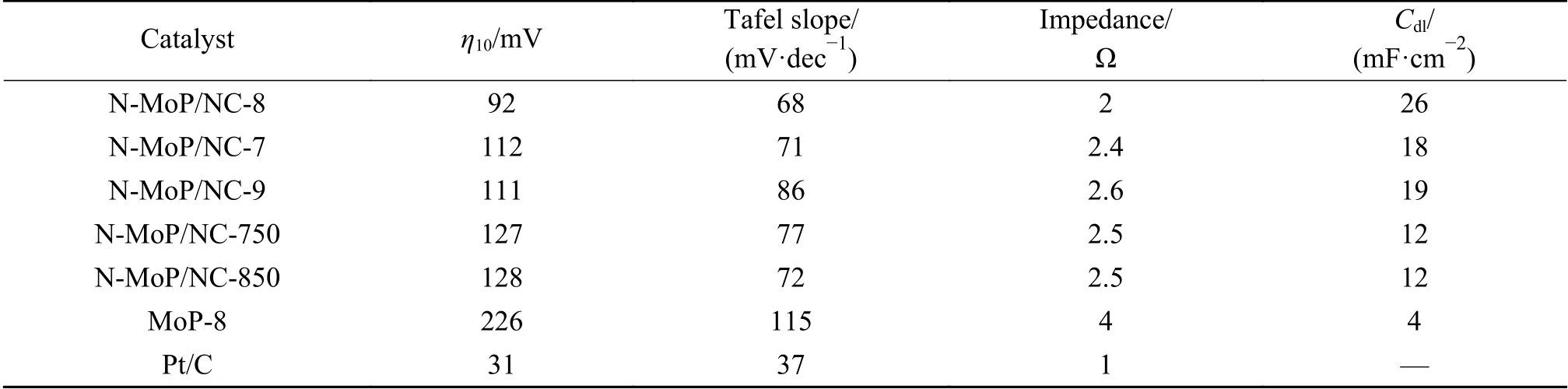
表 4 不同合成条件的MoP和Pt/C的电化学性能Table 4 Electrochemical performance of the MoP materials obtained under different synthesis conditions
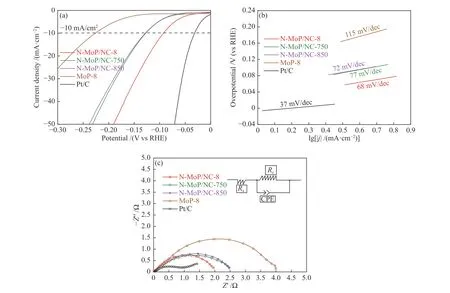
图 7 不同焙烧温度下MoP和Pt/C的 (a) LSV极化曲线,(b) Tafel斜率曲线,(c) EIS曲线Figure 7 LSV polarization curves (a), Tafel slope curves (b), and EIS curves (c) of the MoP materials obtained under different calcination temperatures
固液界面双电层电容(Cdl)与有效电化学活性面积成正比。图8(a)-(f)和表4数据所示,N-MoP/NC-8的Cdl(26 mF/cm2)最大,而其他材料比MoP-8(4 mF/cm2)高了数倍,增加的活性位点与电化学有效活性面积表现一致,进一步证明了TEM和XPS表征得出的结果。到目前为止,合成的N-MoP/NC-8纳米棒催化剂其综合析氢性能优于目前报道的大部分MoP基催化剂,如表5。
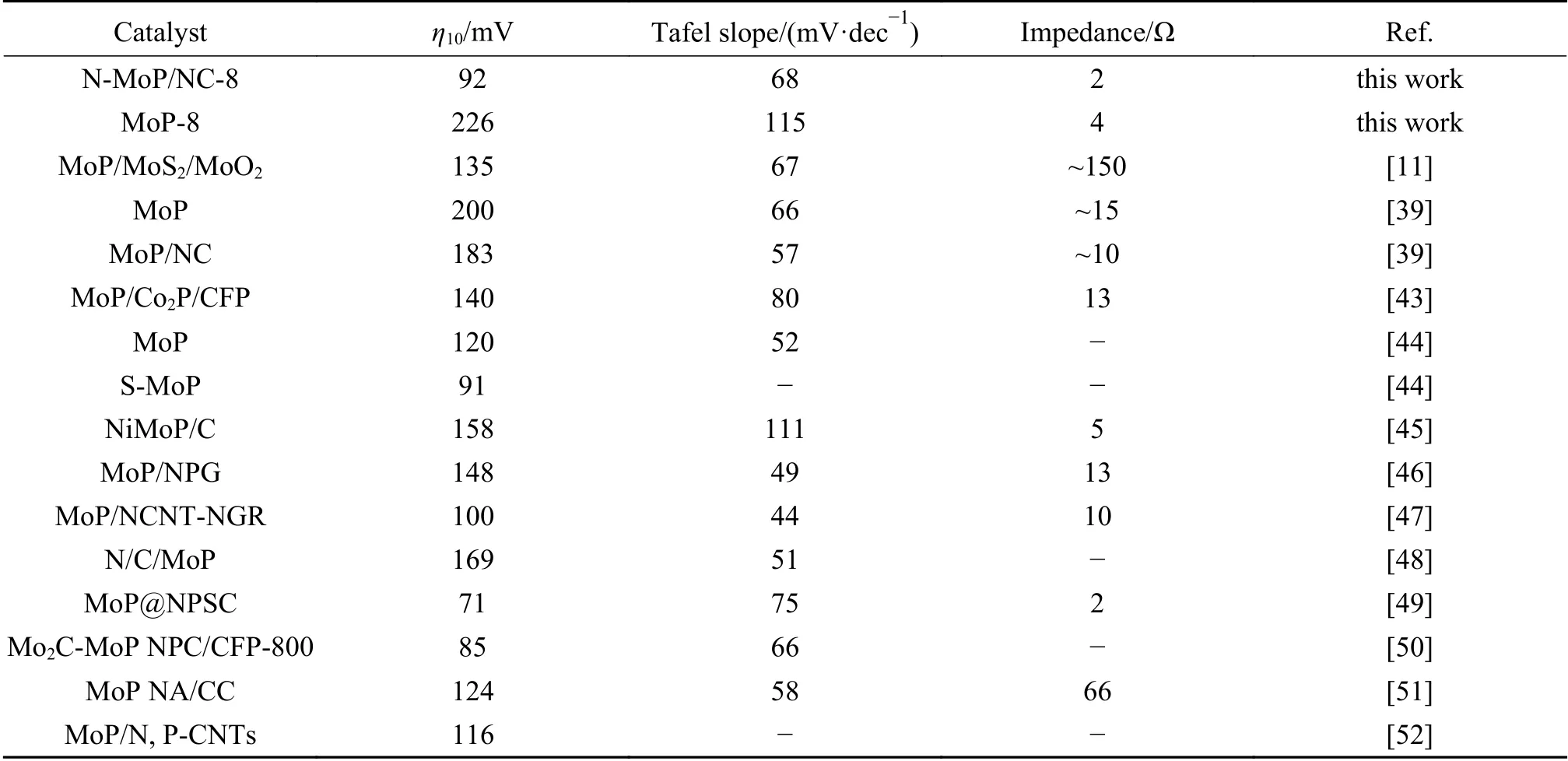
表 5 不同MoP基复合材料的电化学性能Table 5 Electrochemical performance of different MoP-based composites
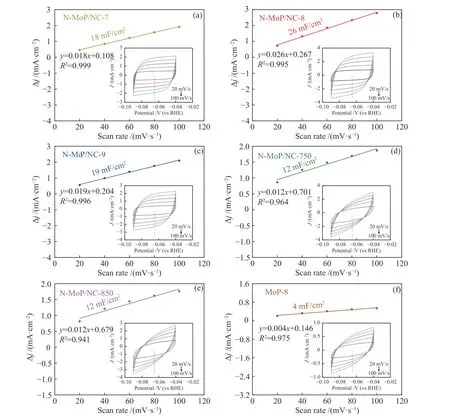
图 8 ((a)-(f)) 不同合成条件的MoP在20-100 mV/s扫描速率下的CV曲线和电容电流曲线Figure 8 CV curves at a scanning rate of 20-100 mV/s and capacitive current curves ((a)-(f)) of the MoP materials obtained under different conditions
催化剂的电化学稳定性也是评估HER性能的重要指标。图9(a)所示,N-MoP/NC-8经过1000次CV后的电流密度只有轻微的降低,过电位的变化可忽略不计,而MoP-8出现了明显的活性损失。同样在20 h的计时稳定电压测试后,N-MoP/NC-8的电位损失相比MoP-8少了近10 mV,证明N-MoP/NC-8比MoP-8具备更高的稳定性。将稳定性测试后的材料进行TEM与XPS表征,结果发现,NMoP/NC-8在炭层的保护下,形貌没有发生明显改变(图9(b)、(c)),材料的氧化态略有增加,但MoP活性相的P-Mo键变化不明显(图9(f)、(g))。相比之下,MoP-8出现许多被酸腐蚀的孔(图9(d)、(e)),P-Mo键损失较为严重(图9(f)、(g)),原因在于炭层可以防止MoP在酸性溶液中被腐蚀,说明炭层提高了材料抗酸能力,有助于稳定性提升。
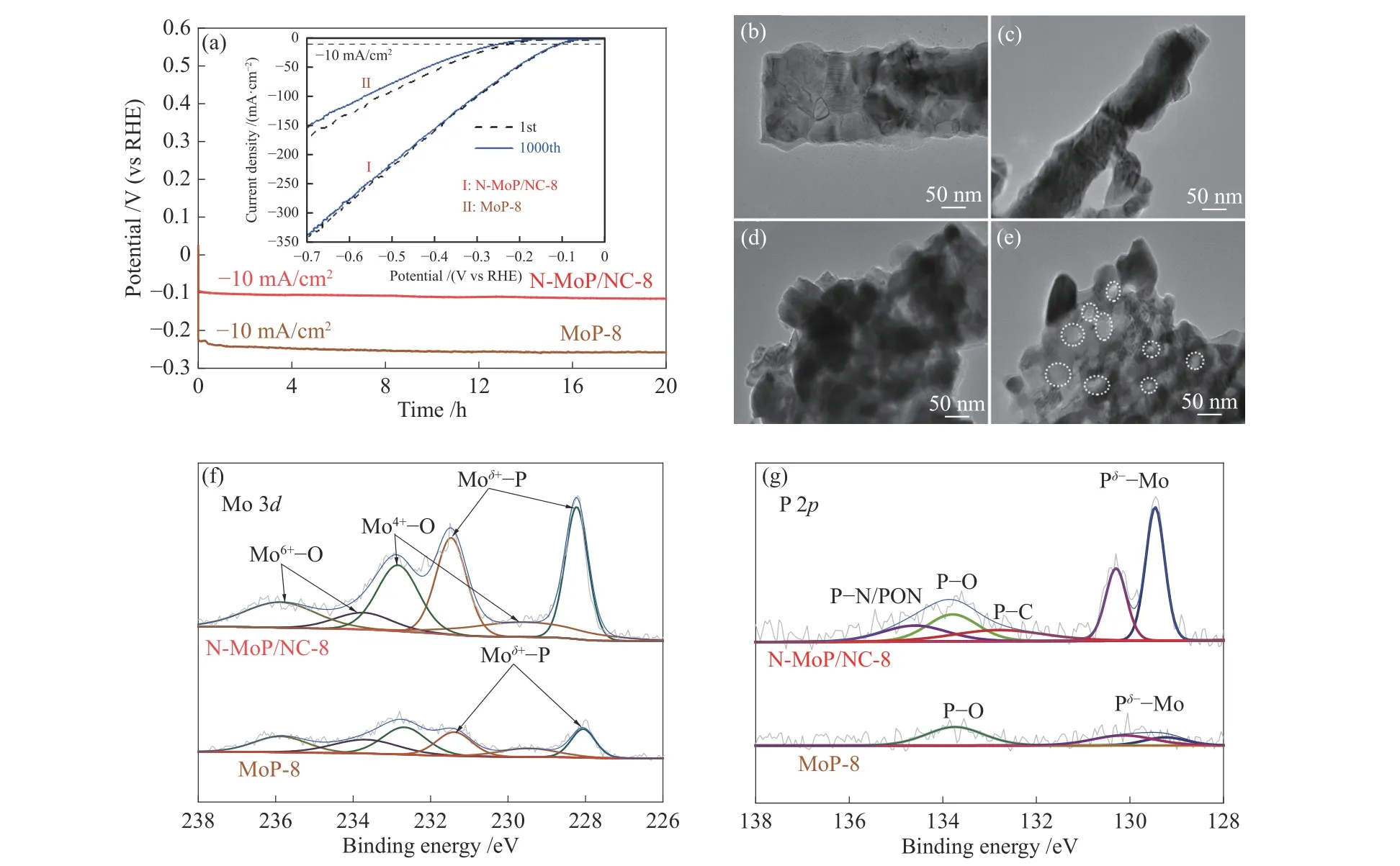
图 9 (a) N-MoP/NC-8和MoP-8的1000圈CV循环前后的LSV极化曲线和计时电位(CP)曲线;((b)、(c)) N-MoP/NC-8和((d)、(e)) MoP-8在CP测试前后的TEM照片;(f) N-MoP/NC-8和 (g) MoP-8在CP测试后的Mo、P元素的XPS光谱谱图Figure 9 LSV polarization curves (a) before and after 1000 cycles of CV and time-dependent potential (CP) curves of N-MoP/NC-8 and MoP-8; TEM images of N-MoP/NC-8 ((b), (c)) and MoP-8 ((d), (e)) before and after CP; Mo and P XPS spectra of (f) N-MoP/NC-8 and (g) MoP-8 after CP
综上,经过改性的N-MoP/NC-8纳米棒相比于MoP-8片状复合材料在过电位、Tafel斜率、阻抗、稳定性等方面展现出更优秀的析氢性能,主要归因于以下几个方面:首先,乙二胺炭化形成了具有炭包覆层的核壳结构,有效限制MoP的烧结团聚,增加了材料比表面积和孔体积,促进更多表面活性位点与H+接触。其次,掺杂在MoP中的N原子会改变周围原子的电子密度,降低Mo的d带中心,调节Hads的吸附-脱附过程来加快HER的析氢反应动力学。另外,掺杂在炭中的特殊N原子引发了材料结构缺陷,部分N原子成为了新的析氢位点,增加了电化学有效活性位点数量。最后,石墨化的炭层为材料提供更有效的电荷转移能力,降低阻抗,提升析氢反应工作效率。同时炭层作为“外壳”与MoP的强作用力复合确保了材料整体抗酸能力和结构稳定,使催化剂具有良好的析氢稳定性。
基于以上原因,N-MoP/NC-8利用了炭层和杂原子掺杂之间的共同调节作用,同时较大的比表面积和孔体积保证催化反应的进行,使其不断向对HER有利的方向发展。
3 结 论
以三氧化钼-乙二胺有机无机杂化材料为前驱体,通过气-固反应合成了将N掺杂MoP封装在炭层的核壳纳米棒催化剂。乙二胺作为提供氮源和碳源的“中介”,辅助MoP原位生长以实现两相复合,控制MoP形态结构,同时引入的N原子对MoP的电子结构进行调节,使催化剂在酸性析氢反应中表现出较好的性能(在10 mA/cm2电流密度下,η10= 92 mV,Tafel斜率为68 mV/dec),良好的电荷转移能力和至少20 h的稳定性。这种利用限域空间效应进行材料两相原位生长同时引入原子掺杂的策略,为未来过渡金属析氢催化剂的制备及改性方向上提供了独特的思路。
猜你喜欢
涂料工业(2022年9期)2022-10-26
涂料工业(2022年9期)2022-10-26
中国塑料(2022年10期)2022-10-26
含能材料(2022年4期)2022-04-16
涂料工业(2022年2期)2022-04-01
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
表面工程与再制造(2019年6期)2019-08-24
新疆医科大学学报(2018年5期)2018-06-11
资源节约与环保(2018年1期)2018-02-08
