
微波消解-原子荧光光谱法测定中药口服液中的砷和汞
2022-12-12 08:53周红梅李兴根
泰州职业技术学院学报 2022年5期
周红梅,李兴根
(1.南京医科大学附属泰州人民医院;2.泰州市产品质量监督检验院,江苏 泰州 225300)
工业化对大气、水体、土壤的污染,造成中药材在栽培种植过程中可能受到不同程度重金属的污染,同时中药传统的加工工艺也会带来一定的污染,从而影响中药的安全性。其中,砷、汞是中药材超标最严重的两种重金属元素,对人体危害极大。2020年版中国药典对多味中药材都增加了砷(As)、汞(Hg)检查的内容。口服液剂是以中药汤剂为基础,提取药物中有效成分,加入矫味剂、抑菌剂等附加剂,并按注射剂安瓿灌封处理工艺,制成的一种无菌或半无菌的口服液体制剂,目前有关口服液中As、Hg 的检测多参照的是中药材和食品的检测方法[1-3],如电感耦合等离子体质谱法,该方法测定砷和汞元素存在干扰,线性范围较窄,汞元素测定时溶液浓度必须控制在2μg/L以下,且检测过程中溶液会残留于仪器进样系统中即记忆效应,试验结果不准确,尚缺乏灵敏度高、专属性强的检测手段,因此建立科学系统的检测方法进行监控非常重要。原子荧光光谱技术在测定中药材中As、Hg 等重金属含量方面具有独特的优势[4],相比电感耦合等离子体质谱法,原子荧光光谱分析技术的校正曲线线性范围更宽,砷和汞溶液浓度可测定至10μg/L 甚至更高,仪器检测过程中基本不会有记忆效应,检测结果重现性好,能同时进行砷和汞两个元素的含量测定。本实验拟以中药口服液(蒲地蓝、蓝岑等)为例,采用微波消解法处理样品,建立原子荧光光谱分析技术测定其中重金属As、Hg 含量的方法。
1 实验部分
1.1 仪器AFS-933 型原子荧光光度计(北京吉天仪器有限公司);Multiwave PRO 微波反应系统(安东帕集团);中央纯水系统(法国威立雅公司)。
1.2 试剂As 标准溶液1000g/mL,国家标准物质研究中心;Hg 标准溶液1000g/mL,国家标准物质研究中心;KBH4溶液1.5%:称取0.5g KOH 溶于100mL 水中,再称取1.5g KBH4溶于此溶液中,混匀;浓硝酸(65%),优级纯,德国默克;浓硫酸(98%),优级纯,国药集团化学试剂有限公司;浓盐酸(37%),优级纯,德国默克;30%H2O2,优级纯,国药集团化学试剂有限公司;试剂为分析纯,水为高纯水。
1.3 样品消解
1.3.1 湿法消解法 称取3mL 口服液试样置于250mL 凯氏烧瓶中,加入10mL 硝酸,放置30 分钟后,缓缓加热,待作用缓和后,稍冷,沿瓶壁加入5mL 硫酸,再缓缓加热,至瓶中溶液开始变成棕色,不断滴加硝酸,至样品完全分解,继续加热,生成大量的二氧化硫白色烟雾,最后溶液应为无色或微带黄色。冷却后加入20mL 水煮沸,除去残余的硝酸至产生白烟为止。如此处理两次,放冷,将溶液移入25mL容量瓶中,加水定容至刻度,混匀备用。同时作试剂空白。
1.3.2 微波消解法 称取3mL 试样于消解罐中,加入10mL HNO3,加盖密闭,同上述步骤制备样品空白。将样品与空白同时置于微波消解仪中,按表1消解程序消解。

表1 微波消解程序
消解程序完成后,冷却30min,缓慢打开消解罐。注意控制开罐速度,防止消解液喷溅造成损失。将消解罐置于电热板上加热赶酸,继续加热浓缩至消解液剩余约2mL,转移至25mL 容量瓶中,用1%硝酸溶液定容至刻度,得到样品测定溶液。同上述步骤制备样品空白。
1.4 试样测定分别吸取消解完全的样品消解液进入各仪器进行测定,根据所测荧光强度利用回归方程计算样品中砷和汞的含量。
2 结果与讨论
2.1 前处理条件的选择移取相同体积(3mL)的口服液样品分别采用湿法消解法和微波消解法对其进行消解前处理,湿法消解法对样品进行前处理后样品仍呈现黄色,消解并不彻底,而微波消解法对样品进行前处理后样品呈现无色透明状态,消解非常彻底。同时,湿法消解法为敞口消解,与密闭消解的微波消解法相比,其在前处理过程中易受到污染,且砷、汞元素均为易挥发元素,采用湿法消解时也极易损失,导致检测结果偏低。因此,选择微波消解法为口服液元素分析的前处理方法。
2.2 标准曲线绘制使用单标移液管准确吸取一定量的砷和汞标准溶液,用1%HNO3溶液作为稀释剂,逐级稀释,分别配制浓度为0.0、2.0、4.0、6.0、8.0、10.0μg/L 的标准工作溶液。分别吸取上面配制的各标准使用液采用原子荧光光度计进行测定,测得其吸光度并求得荧光强度与浓度关系的一元线性回归方程(见图1和图2)。
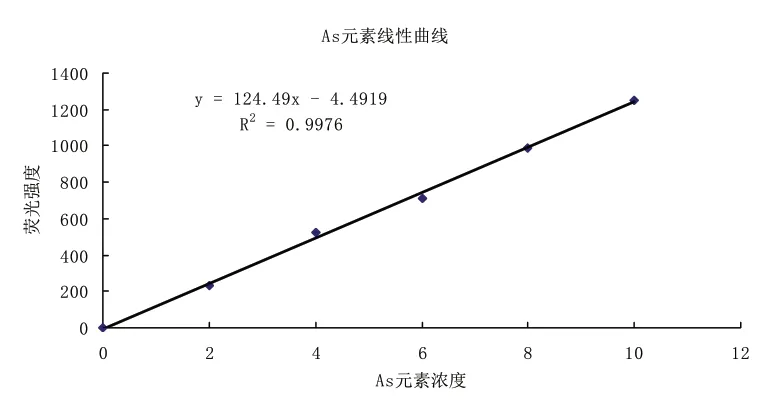
图1 As元素标准曲线
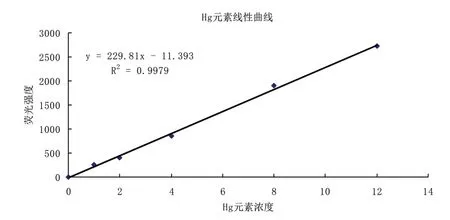
图2 Hg元素标准曲线
2.3 方法检出限和定量限取试剂空白进行11 次平行测定,以其测得的标准偏差3 倍计算,砷的检出限为0.0099μg/L,汞的检出限为0.042μg/L;以其测得的标准偏差10 倍计算,砷的检出限为0.03μg/L,汞的检出限为0.13μg/L。同时在定量限附近平行测定6个样本(参照标准溶液配制),砷的标准偏差为4.52%,汞的标准偏差为5.22%。
2.4 方法的精密度使用该方法对口服液样品进行6次平行测定(见表2和表3)。
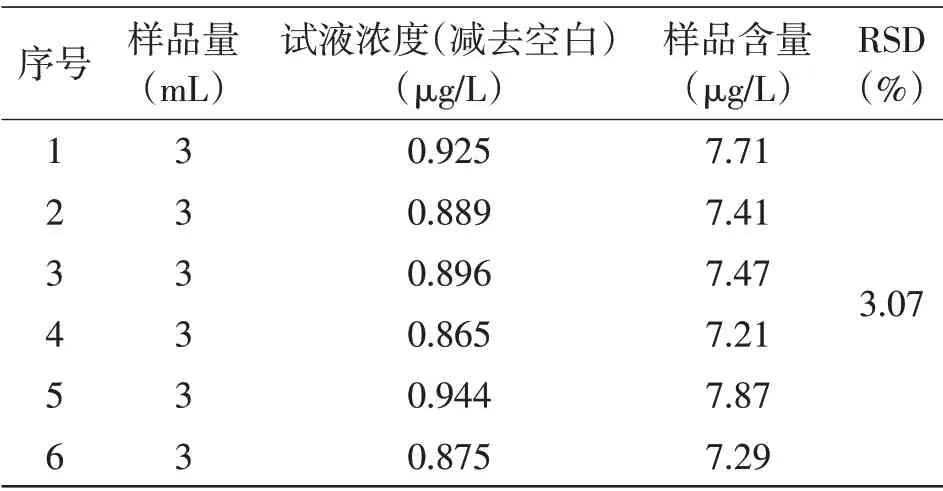
表2 As元素精密度测定
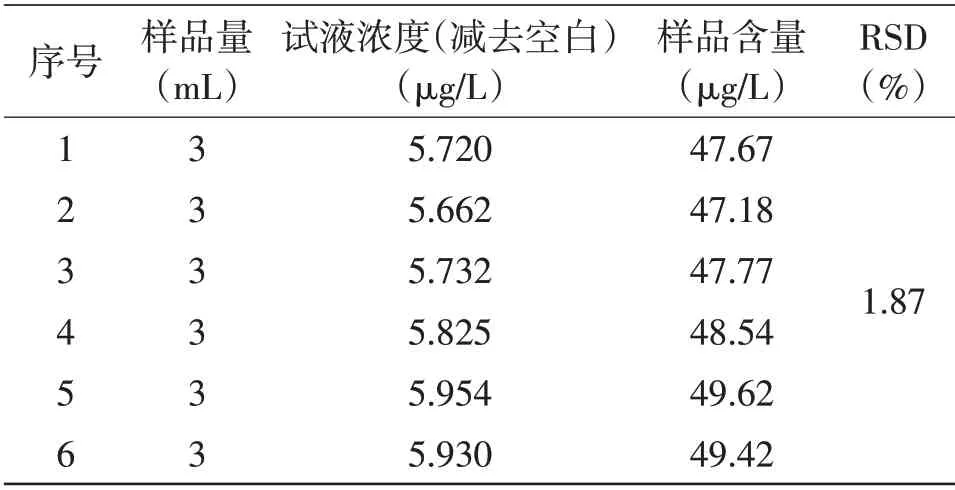
表3 Hg元素精密度测定
2.5 方法回收率试验本方法采用加标回收试验来验证方法的准确性。在口服液样品中加入三个浓度的重金属对照,低浓度加入定量限附近的量,中浓度加入量至总量为质量标准中的限度附近,高浓度加入量至总量为限度的120%,结果见表4、表5。
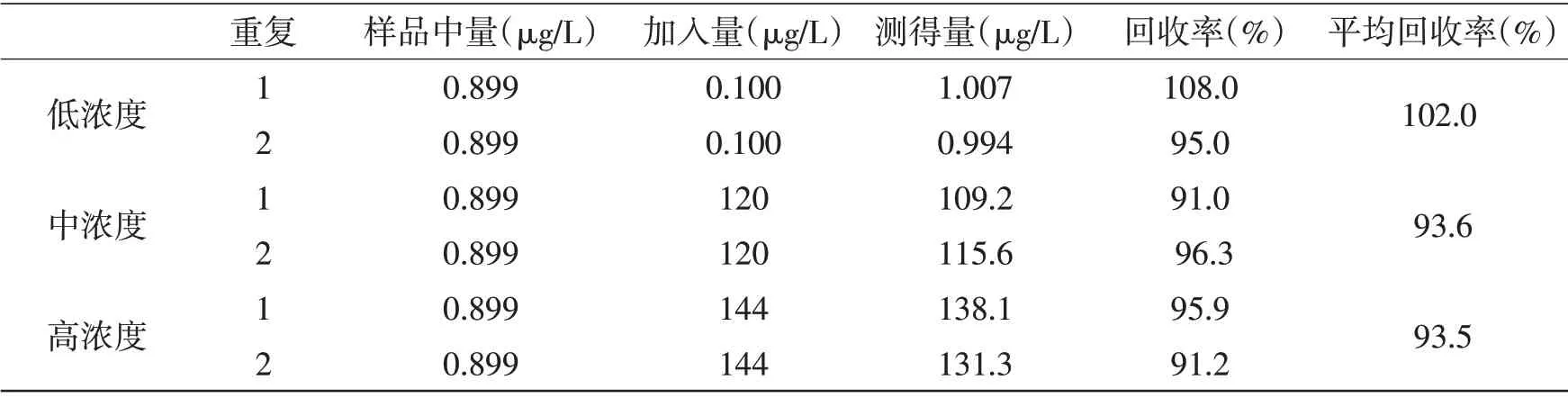
表4 As元素准确度测定
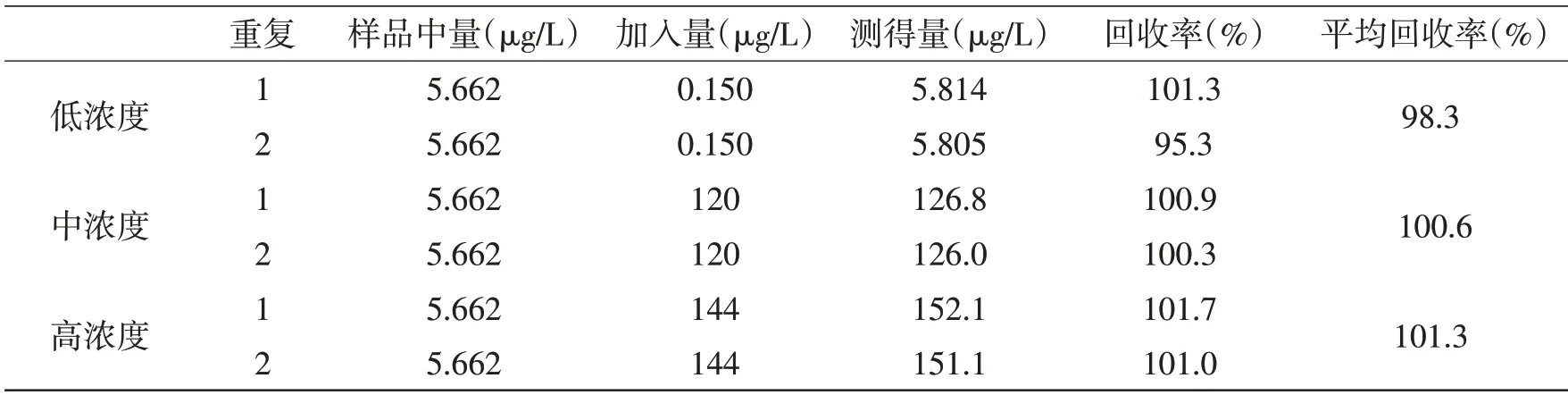
表5 Hg元素准确度测定
2.6 溶液稳定性最后,对口服液样品中砷、汞两种元素消解液的溶液稳定性进行验证,即将配置好的同一待测样品溶液分别室温下放置若干小时后进行测定,具体结果见表6。结果显示,消解液室温放置下,各重金属测得值呈下降趋势,且1小时内对测定结果的影响(下降均不超过0.7%)在可接受范围之内。
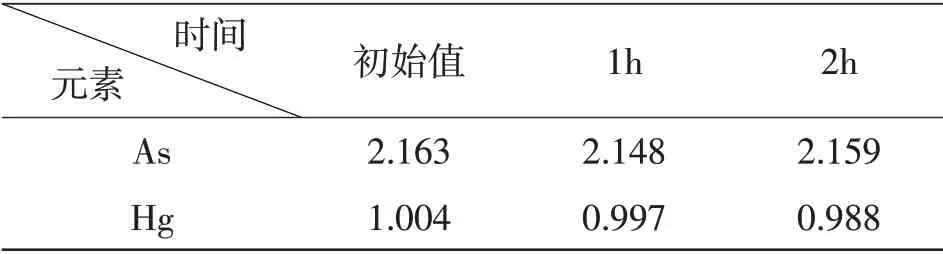
表6 砷、汞元素溶液稳定性验证
3 结论
通过上述的系列试验和实例检测结果可见利用微波消解法测定中药口服液中的砷、汞具有损失低、污染少、试剂利用率高、快速简便的优点,这对满足口服液日常监管品种杂、批次多、要求检验周期短具有重要意义。
猜你喜欢
纺织科学与工程学报(2020年1期)2020-06-12
中成药(2018年1期)2018-02-02
中成药(2017年8期)2017-11-22
中成药(2017年3期)2017-05-17
光学精密工程(2016年2期)2016-11-07
中成药(2016年8期)2016-05-17
中国民族医药杂志(2016年9期)2016-05-09
食品界(2016年4期)2016-02-27
铜业工程(2015年4期)2015-12-29
中国药业(2014年24期)2014-05-26
