
高比表面积羟基硅酸镁的制备及其形成机理研究
2022-12-12 03:05王琪浩王余莲朱益斌刘珈伊时天骄林永瑾田伊笛苏德生袁志刚
沈阳理工大学学报 2022年6期
王琪浩,王余莲,王 楠,李 闯,张 俊,朱益斌,刘珈伊,时天骄,林永瑾,田伊笛,苏德生,袁志刚
(1.沈阳理工大学材料科学与工程学院,沈阳110159;2.太原理工大学材料科学与工程学院,太原 030024;3.辽宁省超高功率石墨电极材料专业技术创新中心,辽宁丹东 118100)
硅酸镁理论分子式为xMgO·ySiO2·nH2O,人工合成的硅酸镁主要有三硅酸镁和六硅酸镁,因其具有比表面积大、吸附能力强等优点,在医药、煎炸油处理、废水处理和生物柴油精炼等领域[1-5]具有广阔的应用前景,如三硅酸镁作为抗酸药和抗癫痫药物的组成部分,已被广泛使用[6],特别是硅酸镁对放射性元素铀、锶具有良好的吸附性能[7-8],在封装放射性核废物领域具有巨大潜质。
硅酸镁的制备方法主要有沉淀法、水热法、溶剂热法、模板法等。 Aysa-Martínez Y 等[9]利用沉淀法制备了SiO2与MgO 摩尔比不同的无定型硅酸镁,其Mg2+利用率接近100%,比表面积为662m2/g。 Sun Z W 等[10]利用水热法和冷冻干燥法制备了多孔分层结构的硅酸镁,比表面积和总孔容分别为530m2/g 和0.90cm3/g,其对亚甲基蓝最大吸附容量达到526mg/g,远高于常规吸附量。 黄人瑶[11]利用成核/水热晶化隔离法制备了硅酸镁,相较于传统水热法,比表面积提高了121%,约为597m2/g,其对Pb2+和Zn2+的吸附容量分别为142.1mg/g 和46.5mg/g,提高了37.3%和25.3%。 程昊等[12]以微米级硅胶球为硅源,将水热合成法和模板法相结合,制备了球形多孔硅酸镁。 Zhao R 等[13]以二氧化硅纤维薄膜为硅源、氨水为沉淀剂,采用水热反应制备了硅酸镁纤维薄膜,其柔韧、坚固,具有较高的抗拉强度和良好的机械稳定性,比表面积为463.4m2/g,对阳离子染料亚甲基蓝的吸附量达到609. 75mg/g。Sun Z W等[14]利用水热法制备硅酸镁/碳复合材料,该材料由无定型硅酸镁和无定型水热煤组成,具有分层多孔结构,其对罗丹明B 平衡吸附容量为244mg/g, 比硅酸镁材料的吸附容量高27.48%。 王学凯[15]利用水热法在硅藻土表面生长出具有独特蜂窝状结构的羟基硅酸镁(Mg3Si4O10(OH)2),其比表面积为149m2/g,对Cr6+的最大吸附容量达到535mg/g。
综上可知,高比表面积羟基硅酸镁制备过程中原料和初始条件改变对其形貌和性能影响显著,且制备工艺均比较繁琐,多孔结构形成机理的研究尚显欠缺。 本文选取Na2SiO3·5H2O 和MgCl2·6H2O为原料,采用沉淀法制备羟基硅酸镁,系统研究硅镁比(反应体系中硅原子和镁原子的物质的量比)、体系pH 值、反应温度、反应时间、煅烧或水热处理对羟基硅酸镁比表面积的影响,并初步分析羟基硅酸镁的形成机理。
1 实验部分
1.1 主要试剂
五水合硅酸钠(Na2SiO3·5H2O)、六水氯化镁(MgCl2·6H2O)、氢氧化钠(NaOH)、盐酸(HCl),均为分析纯,天津市大茂化学试剂厂;去离子水,自制。
1.2 主要设备
DF-101S 集热式恒温加热磁力搅拌器,巩义市予华仪器有限公司;SHZ-D(Ⅲ)循环水式多用真空泵,常州亚旺仪器有限公司;DHG-9076A 电热恒温鼓风干燥箱,上海精宏实验设备有限公司;1400-40HMF 马弗炉,上海皓越电炉技术有限公司。
1.3 样品制备
分别称取一定量的Na2SiO3·5H2O 和MgCl2·6H2O,配制一定浓度的硅酸钠溶液和氯化镁溶液;将硅酸钠溶液滴加到氯化镁溶液中,在35 ~85℃下恒温搅拌,得到混合溶液;采用盐酸或氢氧化钠将上述混合溶液pH 值调节至8 ±0. 05 ~12 ±0.05,反应20 ~140min;将部分混合液体过滤、洗涤、沉淀、干燥、研磨,得到白色粉末,之后在一定温度(300 ~700℃)下煅烧4h,获得煅烧后样品;其余部分混合溶液装入内胆为聚四氟乙烯的反应釜中进行170℃水热处理(0 ~10h),再过滤、洗涤、沉淀、干燥、研磨,得到水热后样品。
1.4 测试与表征
采用日本Rigaku 公司UltimaⅣ型X 射线衍射仪(XRD)对产物的物相组成进行分析,扫描角度5 ~90°,扫描速率10°/min;利用美国康塔公司NOVA 1000e 全自动比表面积分析仪测定样品的比表面积;采用美国FEI 公司Nova 450 型场发射扫描电子显微镜(SEM)表征样品的微观形貌;采用美国热电公司Nicolet380 型傅里叶变换红外光谱仪(FT-IR)分析样品官能团。
2 结果与讨论
2.1 硅镁比对羟基硅酸镁性能的影响
采用反应温度为65℃、反应时间为80min、反应体系pH 值为12 ±0.05,在不同硅镁比下制备羟基硅酸镁,测试其XRD 衍射图谱如图1所示。

图1 不同硅镁比下制备产物的XRD 图谱
由图1可以看出,不同硅镁比下制备产物在衍射角为26°、35°、60°处均呈现出宽化弥散衍射峰,且均与羟基硅酸镁的标准峰(JCPDS 22-1162)对应,表明产物为非晶态羟基硅酸镁[16-18],其组成与α-叶蛇纹石类似[19-20]。 由此说明采用沉淀法可以制得羟基硅酸镁,但结晶度较差,且随着硅镁比增加,羟基硅酸镁结晶度无明显提高。
不同硅镁比下制备产物的比表面积变化曲线如图2所示。
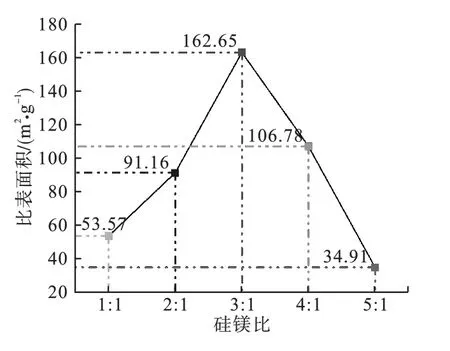
图2 不同硅镁比下制备产物的比表面积变化曲线
由图2可见,随着硅镁比增大,羟基硅酸镁的比表面积先增大后减小;硅镁比为3∶1 时,产物比表面积达到最大值162.65m2/g。 这可能是由于硅镁比较小时,SiO23-较少,阻碍了SiO2网格生长;随着硅镁比增大,Mg2+所占比例减少,SiO2网格生长加快,比表面积增大;硅镁比继续增大,硅酸钠过量加入,则起到粘结剂的作用,反而使比表面积减小。 故选择适宜的硅镁比为3∶1。
2.2 体系pH 值对羟基硅酸镁性能的影响
采用硅镁比为3∶1、反应温度为65℃、反应时间为80min,在不同体系pH 值下制备羟基硅酸镁,测试其XRD 衍射图谱如图3所示。
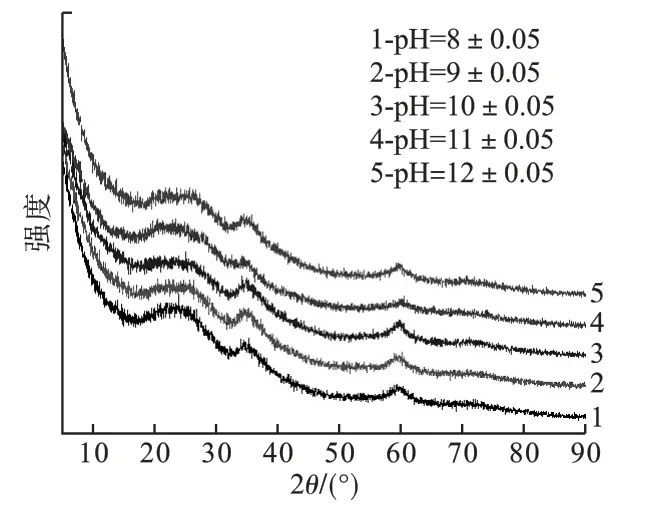
图3 不同体系pH 值下制备产物的XRD 图谱
由图3可知,不同体系pH 值下制得的产物在衍射角为26°、35°、60°处均呈现出宽化弥散衍射峰,且均与羟基硅酸镁的标准峰(JCPDS 22-1162)对应,表明产物为非晶态羟基硅酸镁,其组成与α-叶蛇纹石类似。 随着反应体系pH 值增大,衍射角为19 ~22°处出现凸起,但未形成衍射峰;由于大量OH-和Mg2+作用后与硅烷醇基团(Si—O—H) 反应,形成Si—O—Mg—OH 基团,从而使XRD 衍射图谱上出现新宽峰。 说明调节体系pH值可提高羟基硅酸镁的结晶度。
不同体系pH 值下制备产物的比表面积变化曲线如图4所示。

图4 不同体系pH 值下制备产物的比表面积变化曲线
由图4可见,随着体系pH 值增大,产物比表面积先增大后减小再增大;体系pH 值为10 ±0.05时产物的比表面积最大,达到242.15m2/g。这可能是由于pH 值增大,大量OH-使羟基硅酸镁表面形成更多硅烷醇基团(Si—O—H)和Si—O—Mg—OH 基团,从而使比表面积增大;但过量OH-影响Mg2+、SiO23-的水解平衡,使羟基硅酸镁内部的Mg2+析出,与OH-形成Mg(OH)2胶体或沉淀,导致表面结构破坏引起部分孔道堵塞,比表面积骤减;当pH 值为12 ±0.05 时,羟基硅酸镁内部结构进一步破坏,反而使部分孔道开放,导致比表面积略有上升。 故选择适宜的反应体系pH值为10 ±0.05。
2.3 反应时间对羟基硅酸镁性能的影响
采用体系pH 值为10 ±0.05、硅镁比为3∶1、反应温度为65℃,在不同反应时间下制备羟基硅酸镁,测试其XRD 衍射图谱如图5所示。
由图5可知,不同反应时间下制备的产物在衍射角为26°、35°、60°处均呈现宽化弥散衍射峰,且均与羟基硅酸镁的标准峰(JCPDS 22-1162)对应,表明产物为非晶态羟基硅酸镁,其组成与α-叶蛇纹石类似。 当反应时间为100min 时,衍射角为19 ~22°处出现凸起,说明反应时间在一定程度上影响羟基硅酸镁的结晶度。

图5 不同反应时间下制备产物的XRD 图谱
不同反应时间下制备产物的比表面积变化曲线如图6所示。

图6 不同反应时间下制备产物的比表面积变化曲线
由图6可见,随着反应时间延长,产物比表面积先增大后减小再增大,之后再略有减小。 反应时间为80min 时,产物比表面积最大, 达到242.15m2/g。 反应时间在20 ~80min 之间时,比表面积急剧上升,可能由于羟基硅酸镁内部[SiO4]四面体和硅氧烷基团(Si—O—Si)生长、表面Si—O—Mg—OH 基团形成所致;反应时间增加至100min 时,由于羟基硅酸镁对反应物与生成物的吸附,可能导致孔道堵塞,比表面积有所减小;反应时间继续延长,堵塞孔道的部分反应物继续反应,使羟基硅酸镁比表面积略有增加;之后再延长反应时间,比表面积小幅下降。 故选择适宜的反应时间为80min。
2.4 反应温度对羟基硅酸镁性能的影响
采用反应时间为80min、体系pH 值为10 ±0.05、硅镁比为3∶1,在不同反应温度下制备羟基硅酸镁,测试其XRD 衍射图谱如图7所示。
由图7可知,不同反应温度下制备产物均为非晶态羟基硅酸镁;反应温度为85℃时,衍射角为26°处宽峰较为明显,说明适宜反应温度可使羟基硅酸镁的结晶度提高。

图7 不同反应温度下制备产物的XRD 图谱
不同反应温度下制备产物的比表面积变化曲线如图8所示。

图8 不同反应温度下制备产物的比表面积变化曲线
由图8可见,随着反应温度提高,产物比表面积先增大后减小。 反应温度在35 ~75℃之间时,随着反应温度提高,硅酸镁层状结构中孔道被打开,比表面积增大;反应温度为75℃时产物比表面积最大,达到263.37m2/g;当反应温度超过75℃时,反应液中SiO23-缩聚增强,导致[SiO4]四面体层间连接程度提高,形成更为紧密的结构,故比表面积减小。 因此选择适宜的反应温度为75℃。
2.5 煅烧温度对羟基硅酸镁性能的影响
对上述适宜条件下制备的产物于不同温度下煅烧4h,得到煅烧后羟基硅酸镁,测试其XRD 图谱,如图9所示。
由图9可见,不同煅烧温度下制备产物的衍射峰与羟基硅酸镁的标准峰(JCPDS 22-1162)对应,表明产物为非晶态羟基硅酸镁。 煅烧温度为500℃时,衍射角为26°处的衍射宽峰高度明显下降;煅烧温度为600℃时,衍射角为35°和60°处的衍射峰高度下降;煅烧温度为700℃时,产物在衍射角为35°和60°处的衍射峰完全消失,且第一个宽峰更加突出。 由此说明产物在450℃以下煅烧时,其结晶度及物相组成无明显变化;在450 ~600℃煅烧时,羟基硅酸镁内部结构被破坏;在600℃以上煅烧时,羟基硅酸镁内部结构则被完全破坏,孔结构坍塌,会导致比表面积骤降。

图9 不同煅烧温度下所得产物的XRD 图谱
不同煅烧温度下制备产物的比表面积变化曲线如图10 所示。

图10 不同煅烧温度下制备产物的比表面积变化曲线
由图10 可知,随着煅烧温度升高,产物比表面积先增大后减小;煅烧温度为400℃时,所得产物的比表面积最大,达到336.16m2/g;煅烧温度为700℃时,产物比表面积小于未煅烧产物。 这是由于煅烧温度小于400℃时,羟基硅酸镁中结晶水被除去,孔道打开使比表面积增大;煅烧温度大于400℃时,结合XRD 分析结果可知,羟基硅酸镁内部结构可能重排,随着温度升高,结构重排加剧,导致比表面积减小,最后结构破坏。 故适宜的煅烧温度为400℃。
2.6 水热时间对羟基硅酸镁性能的影响
对上述适宜条件下制备的产物(未经煅烧)在170℃下进行不同时间的水热处理,测试得到其XRD 图谱,如图11 所示。
由图11 可见,各衍射峰与羟基硅酸镁标准图谱(JCPDS 22-1162)一致,表明产物为非晶态羟基硅酸镁。 水热处理后样品在衍射角为20°处的衍射峰变得尖锐,说明经过水热处理,羟基硅酸镁结晶度提高。

图11 不同水热时间下制备产物的XRD 图谱
不同水热时间下制备产物的比表面积变化曲线如图12 所示。

图12 不同水热时间下制备产物的比表面积变化曲线
由图12 可见,随着水热时间延长,产物比表面积先增大后减小;水热时间为7h 时,产物比表面积最大,达到425.30m2/g,与未水热处理样品(图8)相比增加61.48%。 其原因可能是水热处理使得羟基硅酸镁结晶度提高且团聚减少,故比表面积增大;但水热时间过长时,具有较高比表面积和结晶度的样品活性也较高,且水热釜压力较大,其对反应物和产物的吸附作用也增强,使得团聚增加,比表面积减小。
由煅烧和水热处理结果可知,样品水热处理后性能优于煅烧处理后性能,故优先选择水热处理且水热处理时间为7h。
170℃水热处理前后制备产物的FT-IR 谱线如图13 所示。

图13 170℃水热处理前后制备产物的FT-IR 谱图
由图13 可知,170℃水热前后所得产物均在波数为3420 ~3460cm-1、 1630 ~1640cm-1、1 010cm-1、905 ~907cm-1、652 ~657cm-1、441 ~445cm-1处出现特征峰[21-23]。 特征带3420 ~3460cm-1出现的原因可归为产物表面存在二分子缔合的O—H 基团,1630 ~1640cm-1处特征带是由于—OH 的弯曲振动;特征带1010cm-1出现的原因可归为产物表面存在Si—O—Mg 基团;特征带905 ~907cm-1和441 ~445cm-1的出现是因为产物表面具有Si—O—H 基团;出现特征带652 ~657cm-1原因可归为产物表面具有Mg—OH 基团。 由前述XRD 分析结果可知,产物中含有Si、O、Mg、H 元素,与图13 中光谱特征带基团相一致,因此可进一步确定所制备产物为非晶态羟基硅酸镁Mg3Si2O5(OH)4。 由170℃下水热前后所得产物的FT-IR 光谱比对发现,水热处理产物与未水热处理产物的特征带所处位置一致,但水热处理后产物特征带的强度高于水热处理前。
170℃水热处理前后羟基硅酸镁的SEM 图像如图14 所示。
由图14 可以看出,未经水热处理的羟基硅酸镁呈无规则块状,且周围存在聚集颗粒,颗粒间存在空隙;经水热处理后仍呈块状,但其表面存在片层状物,片层相互堆叠形成大面积且不均匀的孔道,由此可解释水热处理后其比表面积骤增的主要原因。


图14 170℃水热处理前后羟基硅酸镁的SEM 图像
综合上述分析,制备羟基硅酸镁的适宜条件为:硅镁比3∶1、体系pH 值10 ±0.05、反应温度75℃、反应时间80min、水热时间7h。 该条件下制备的羟基硅酸镁具有丰富的孔道,比表面积为425.30m2/g。
3 羟基硅酸镁形成机理
羟基硅酸镁属于三方晶系,空间群为P31m(157),其晶格常数a=0.53nm、b=0.918nm、c=0.728nm。 利用Diamond 软件绘制沿b轴[010]和c轴[001-]方向的羟基硅酸镁晶体结构如图15所示。
由图15a 可见,羟基硅酸镁是由[SiO4]四面体与[MgO6]八面体沿(001)方向生长形成的三层状结构,其中[SiO4]四面体为第一层,[MgO6]八面体为第二层,—OH 为第三层,三层组成完整的羟基硅酸镁分子层。 羟基硅酸镁中存在四种不同的桥氧(O1、O2、O3、O4),第一层[SiO4]四面体由O2 原子相互连接;第二层[MgO6]八面体由O3 原子连接;[SiO4]四面体与[MgO6]八面体层间由O1 原子连接;—OH 层与[MgO6]八面体层间由O4 原子连接。由图15 b可见,[SiO4]四面体排列形成六边形空腔,且六边形空腔中存在独立羟基。

图15 羟基硅酸镁晶体结构图
羟基硅酸镁的形成机理示意如图16 所示。
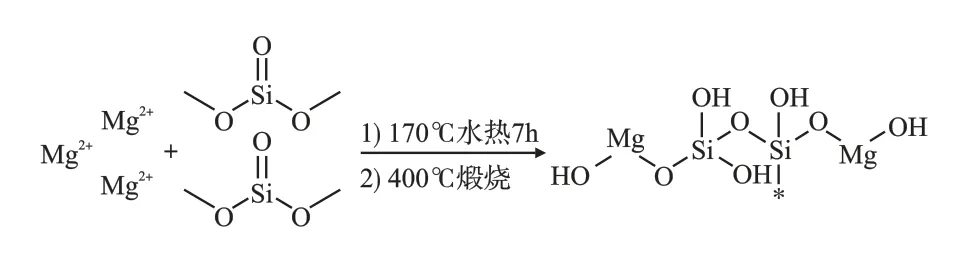
图16 羟基硅酸镁形成机理示意图
羟基硅酸镁的形成主要由于SiO2-3与Mg2+相互作用,经过成核、生长获得。 硅酸盐溶液与镁盐溶液混合后,SiO2-3主要以原硅酸(即H4SiO4)的形式存在。 水热或煅烧条件下,Mg2+与羟基、水分子等通过羟基桥联或氧桥联形成[MgO6]八面体;[SiO4]四面体相互连接形成硅氧烷基团(Si—O—Si);Mg2+、游离的羟基以及硅烷醇基团(Si—O—H)形成Si—O—Mg—OH 基团。 适当pH 值时,溶液中OH-可为Si—O—Mg—OH 基团和硅烷醇基团(Si—O—H)的形成提供羟基,有助于提高硅酸镁表面活性和比表面积[24-25]。
4 结论
(1)适宜的羟基硅酸镁制备条件为:硅镁比3∶1、体系pH 值10 ±0.05、反应温度75℃、反应时间80min、水热时间7h,该条件下可制得比表面积为425.30m2/g 的羟基硅酸镁。
(2)水热处理后羟基硅酸镁结晶度提高,片层状物相互堆叠形成大面积且不均匀的孔道,极大增加了其比表面积。
(3)羟基硅酸镁是由[SiO4]四面体、[MgO6]八面体以及羟基形成的三层状结构。 适当pH 值下,溶液中OH-可为Si—O—Mg—OH 基团和硅烷醇基团(Si—O—H)的形成提供羟基,有助于提高羟基硅酸镁表面活性和增大其比表面积。
猜你喜欢
陶瓷学报(2021年3期)2021-07-22
科学与财富(2021年33期)2021-05-10
陶瓷学报(2019年6期)2019-10-27
陶瓷学报(2019年6期)2019-10-27
新课程·下旬(2019年7期)2019-09-17
汽车零部件(2018年5期)2018-06-13
发明与创新·中学生(2017年11期)2017-12-07
安徽工业大学学报(自然科学版)(2015年4期)2015-12-14
中南大学学报(自然科学版)(2015年1期)2015-09-22
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
