
无定形药物制剂物理稳定性表界面影响机制的研究进展
2022-12-09 09:58:08王亚男刘子莹
食品与药品 2022年6期
王亚男,徐 嘉, 刘子莹,施 秦
(江苏医药职业学院 药学院,江苏 盐城 224005)
口服给药作为临床上最常用的给药方式,具有生产成本低,给药方便及患者顺应性好等优点。近年,随着药物开发手段的进步,分子设计及高通量筛选技术逐步应用到药物的研发过程中,大量新的化合物被开发成为候选药物。但候选药物结构日趋复杂,最新研究显示约80 %的处于研发管线中的候选药物水溶性极差,难以被胃肠道吸收,严重影响其作为口服药物的生物利用度及疗效[1]。众多疗效显著的新药由于水中溶解度低的问题而被迫中止于临床前研究阶段,给其成药性带来严峻挑战。如何有效地递送难溶性药物使其达到理想的口服生物利用度,成为新药成药性研究亟需解决的关键问题之一。
药物的无定形化,是指将长程有序的药物晶体转变为其长程无序的无定形态,是提高难溶性药物溶解度和溶出速率常用的药剂学手段之一。基于该技术,目前已有多种无定形药物及药物-高分子组成的固体分散体被成功开发为市售制剂[2]。但由于无定形药物处于高能态,易于转变为其能量更低的晶态[3],如德国施瓦茨公司于2006年上市的用于治疗早期原发性帕金森病的无定形药物罗替戈丁贴剂在储存和使用过程中出现雪花状的药物结晶,严重影响了药物的递送和治疗效果[4]。该产品于2007年底被欧洲医药管理局强制召回,直到2012年才重新修改处方后上市。尽管在过去的20年间,无定形药物制剂技术已经取得了很大发展,但如何有效维持无定形药物的物理稳定性依然面临着众多挑战。无定形药物在制备、贮存、使用过程中均存在向其晶态转变的风险,会造成溶出降低,甚至严重影响药物发挥治疗效果,依然是相关制剂开发需要面对的重要问题。
尽管围绕无定形药物结晶和物理稳定性研究已取得一定进展,但基于无定形药物整体性质如熵、分子松弛构建的物理稳定性机制解释和预测模型均存在一定缺陷,主要表现为机制解释和预测模型结果与实际情况不符。研究表明表界面性质会显著影响无定形药物制剂的物理稳定性,因此目前该领域的研究重点开始由整体性质研究逐渐转变为对整体性质结合表界面、晶体-无定形交界面等性质共同开展研究[5-6]。此外,高分子材料可对无定形药物物理稳定性和溶出产生显著影响,其对无定形药物的影响机制也是目前的研究热点之一(见图1)。本文总结了近年无定形药物物理稳定性、溶出及过饱和维持等方面的研究进展,重点关注界面和表面的影响机制,旨在为无定形药物制剂的物理稳定性维持和预测提供参考和借鉴。
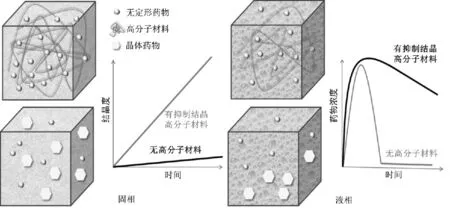
图1 高分子对无定形药物物理稳定性和过饱和维持影响示意图
1 无定形药物成核与表界面影响
无定形药物的结晶可分为成核和结晶生长两步,其中成核是关键的第一步。成核是一个瞬时发生在极小区域内、分子水平上的过程,难以有效进行实时跟踪研究。目前有多种机制用于解释无定形药物的成核过程,主要包括经典和非经典理论[7]。根据经典成核理论,在成核过程中,最先需要少量分子聚集组装成一个团簇,只有团簇达到临界成核尺寸后才能成为真正意义上的晶核,然后结晶生长。根据该理论,影响无定形药物成核的主要因素包括:热力学驱动力,界面自由能,动能和分子识别作用,其具体速率可描述为下式(公式1)。

其中kJ是指分子参与成核过程所需振动频率,Wc代表形成临界尺寸晶核所需克服的热力学能垒。Wc可用下式(公式2)表示。

其中σ表示晶体-流体界面自由能,ΔGv则代表晶体-流体自由能差。目前仍无法通过实验或模拟来获取σ的准确值,但σ的轻微变化会导致成核速率发生数量级的变化。因此,从经典理论衍生出来的成核速率公式(公式1)更多的是充当拟合模型而非预测模型。
经典成核理论研究和应用最为广泛,它可描述不同系统中的动力学和热力学情况,如2019年Bai等[8]围绕水结冰过程开展研究时,发现当形成的冰核大小超过临界尺寸时,水才会开始结冰,该尺寸与经典成核理论预测一致。在药物成核研究领域,Huang等[9]通过一步及两步成核法研究了山梨醇、阿拉伯糖醇、木糖醇和甘油等结构类似药物的成核,发现晶体生长速率相似的条件下,结构类似的多元醇类药物成核速率有显著差异。如图2所示,其成核速率的温度依赖性也符合经典成核理论。Yao等[10]在此基础上研究高分子材料聚维酮(PVP)的添加对多元醇类药物成核的影响,研究表明 PVP 可有效抑制山梨醇和阿拉伯醇成核,且该抑制作用随PVP含量增加及分子量增大而显著增加。Zhang等[11]最新研究表明经典的抗真菌药物氟康唑的成核同样可由经典成核理论进行解释,且其不同晶型成核速率之间呈数量级的差异。该作者指出高分子添加引起的药物分子运动的改变,可能是影响形成临界成核尺寸晶核分子聚集动力学的关键因素[11]。
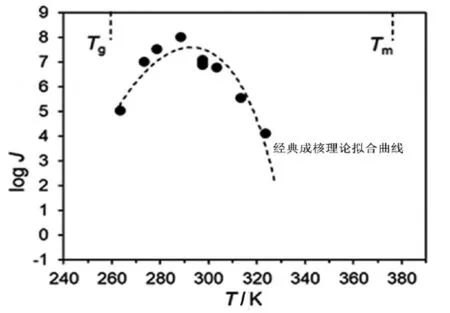
图2 阿拉伯糖醇的成核速率温度依赖性及经典成核理论拟合[9]
近年研究表明表界面是影响药物成核的重要因素之一[12-13]。最新研究表明无定形药物灰黄霉素可在远低于其玻璃化转变温度(Tg)下通过皱缩脆裂效应,形成内部界面结构,可有效加速成核[12]。这种内部界面加速形成的晶型是灰黄霉素亚稳态的II型和III型,在表面自发成核形成的是稳定晶型I型[12]。界面诱导的选择性成核在其他多晶型药物体系中也有类似报道[13]。以经典的非甾体抗炎镇痛药物吲哚美辛为例,在其表面主要观测到热力学稳定的γ晶型及亚稳型的α晶型,其过冷液体内部更多的是亚稳型的δ晶型[12]。除无定形表界面外,晶体界面同样可会成为新的成核位点,其中最经典的是由Yu教授发现并系统研究的“交叉成核”(crossnucleation)现象[14]。该现象最早发现于甘露醇熔融态结晶,此过程中发现其α-晶型可在其原有的δ晶型界面上成核并生长,此时并非发生传统意义上的转晶。目前为止这种晶体界面交叉成核现象已在多个药物和聚合物体系中被观测到[14-16]。在交叉成核中,不同晶型之间的热力学稳定性并非是关键的影响因素。研究表明通过交叉成核形成的新晶型,与原晶型相比通常具有相同或更快的晶体生长速率[6]。因此,对于拥有交叉成核现象的多晶型体系而言,体系在结晶过程中最后形成的晶型,起决定性因素的可能是晶体生长速率而非成核速率。此外,交叉成核现象的出现某种程度上也使常用的添加晶种调控晶型策略无法得到保障。最新研究表明,激光诱导结晶技术中,激光诱导的空化气泡形成的界面可作为药物的成核位点,可有效降低成核能垒,加速部分亚稳晶型成核[17]。随着激光脉冲能量的增加,如磺胺类抗菌药物磺胺噻唑的II型和IV型的成核速率显著增加,导致其最终产物II型和IV型的比例升高[17]。
2 无定形药物晶体生长及表界面影响
与成核相比,无定形药物晶体生长方面的研究相对成熟。在接近熔点(Tm)时,结晶生长主要受热力学驱动力控制,此时温度降低有利于加速晶体生长。当温度远低于Tm时, 晶体生长与药物分子运动的快慢直接相关(研究证实关键控速因素为体相扩散而非黏度[18]),温度降低,分子运动降低,晶体生长变慢。当温度进一步降低[接近或者低于玻璃化转变温度(Tg),部分无定形药物如灰黄霉素[19]、硝苯地平[20]、吲哚美辛[21]等会出现特殊的晶体生长加速作用,该现象被称为玻璃态-晶体(glass-to-crystal,GC)生长。目前,有多种机制被提出用于解释这种特殊的结晶模式,部分机制均涉及到表界面相关性质[3]。如均相成核结晶机制指出GC生长源于晶核在已有晶体界面的富集[22],张力诱导界面动力机制则认为GC生长是由体积收缩产生张力诱导界面分子加速运动所导致[23]。两种机制虽然切入点不同,但都共同考虑到界面性质的重要性。Sun等[24]指出GC生长极有可能是一种固相转变行为,由局部分子运动控制,仅需极小范围内的分子重排即可实现结晶。在该研究基础上,Musumeci等[21]研究数十种具有GC生长的化合物,指出GC生长这种固相转变过程由一个3 nm左右的固-液界面所维持,随温度升高,体系流动性增加,该界面结构被破坏,GC生长终止。最新研究证实GC生长会形成很多小的空隙结构(见图3),这些结构提供分子快速运动所需的界面,从而形成快速的GC生长[25]。

图3 邻三联苯的晶体生长形貌图[25]
近年研究表明无定形药物可在其自由表面发生快速的晶体生长[3,5]。该现象最早发现于非甾体抗炎镇痛药吲哚美辛,随后也在硝苯地平[26]、灰黄霉素[19]、非洛地平[27]、塞来昔布[28]等多个药物中发现。与体相晶体生长相比,表面晶体生长的速率差异可存在数量级差异,如抗真菌药物灰黄霉素在50 ℃(Tg38 ℃)时其表面晶体生长比体相晶体的生长速率快100倍以上[19]。借助表面光栅衰减技术,Yu等[5,29]成功测得多种无定形药物的表面扩散系数。研究证明表面分子运动远快于体相分子运动,两者差异在Tg时达到6~8个数量级,且两者差异随温度降低增大[5]。这可能与表面的特殊环境有关,与体相分子相比,表面分子活动自由度更大[30]。进一步的研究发现分子的表面扩散速率取决于分子间相互作用的强弱及分子本身的大小,相互作用愈强,分子越小,其表面分子运动越快[31-32]。Mirigian等[33]构建基于统计力学的模型来实现对表面分子松弛的预测,在该预测模型中,表面分子快速运动与其周围临近的分子较少有关,同时分子重排所需弹性成本也较低。该模型是建立在分子在表面的嵌入深度与分子自身大小及局部摩擦直接相关这一假设基础上,其所预测的表面分子运动与实测值相符[33]。
对表面晶体生长而言,除表面分子快速运动外,还需要重点关注的是表面晶体生长前沿界面[19,34-35]。Sun等[36]通过原子力显微镜发现药物的表面晶体是高于无定形态表面生长的,其相对高度可达到数十到数百纳米。他们指出这样的生长可充分利用表面快速运动的分子并可有效规避缓慢体相结晶的影响[36]。Hasebe 等[34]发现当温度升高进入过冷液体态区域时,体系流动性的增加会对药物的表面晶体生长产生显著影响。研究证实体系流动性的影响程度与晶体生长微观前沿界面结构密切相关,微观形貌为针状的α-吲哚美辛晶体会被流体严重破坏。但微观结构为块状结晶的γ-吲哚美辛和灰黄霉素I型则可在一定程度上有效抵御流体的破坏作用。Musumeci等[35]通过扫描电子显微镜进一步深入分析了流体对表面晶体生长产生破坏作用的原因。他们发现,当温度升高至Tg以上时,过冷液体会逐渐涌向表面晶体周围,对于块状结晶而言,过冷液体无法有效破坏其晶体-流体交界面,但对于针状晶体而言,过冷流体可从针状晶体的侧面覆盖表面晶体,引起其晶体/流体交界面形貌的巨大变化,从而破坏其生长(见图4)[35]。

图4 流体对吲哚美辛表面不同晶型的影响[35]
除表面外,无定形药物体相中也会在特定条件下形成界面结构,从而加速药物的晶体生长[19,25,37]。无定形药物处于玻璃态易发生脆裂,形成裂纹结构[38]。研究表明药物可在脆裂形成的裂纹结构的孔道中形成快速的晶体生长,该生长速度的快慢与裂纹孔道尺寸正相关[25]。伴随孔道尺寸增加,晶体沿着孔道生长的速率不断加快,并最终与表面晶体生长速度达到一致[25,37]。同样,在过冷液体中,无定形药物结晶导致的体积收缩有时会在体相中引入气泡结构。当晶体在特定温度条件下遇到这些气泡结构时,会诱导形成远快于体相晶体生长的突触状晶体生长。晶体不断推动气泡前进,研究证实气泡诱导晶体生长同样可与表面晶体生长速率一致[19,37]。气泡诱导和裂纹诱导的快速晶体生长均会产生连锁效应。如图5a所示,气泡诱导的晶体生长侧面遇到另一个气泡时,会以另一个气泡为前端诱导出一个新的突触状晶体生长。同样的,当一个裂纹诱导的晶体遇到另外一个裂纹时,晶体会沿着新的裂纹形成新的快速晶体生长(见图5b)。可以想象的是,当无定形样品体相中存在众多的气泡或者裂纹结构时,结晶会沿着这些气泡和裂纹快速生长。这些气泡和裂纹结构提供的自由表面,不仅可加速晶体的快速生长,同时也可成为异相成核的位点,这些界面诱导的结晶会严重影响到无定形药物制剂的物理稳定性。

图5 界面诱导无定形态灰黄霉素快速结晶[37]
3 二元体系晶体生长中高分子界面富集
高分子材料掺杂可显著提高无定形药物物理稳定性,因此无定形药物市售制剂占比较多的是无定形药物分散在高分子聚合物基质中的固体分散体形式[2]。目前,有多种机制用于解释高分子聚合物对无定形药物晶体生长的影响机制,主要包括药物-高分子间相互作用和高分子链段动能机制[39-43]。Kestur等[39]研究一系列高分子对无定形抗高血压药物非洛地平晶体生长的影响,发现高分子材料与药物之间相互作用越强,其对晶体生长的抑制作用愈明显。Kothari等[41]指出药物-高分子材料的相互作用可显著降低无定形体系的整体分子运动,是导致晶体生长显著降低的重要原因。Powell等[42]在研究中却发现药物-高分子材料的相互作用并非是影响二元体系药物晶体生长的关键因素,高分子聚合物本身的链段动能是药物晶体生长的控制因素。在随后的研究中,Huang等[43]指出高分子材料极有可能在药物晶体生长过程中在晶体生长前沿富集,影响穿越该区域的药物分子运动,从而调控晶体生长(见图6)。
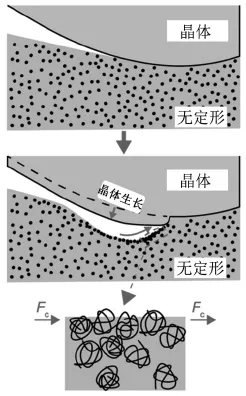
图6 二元体系无定形药物晶体生长高分子前沿富集示意图[43]
Zhang等[44]研究发现高分子聚合物对伊曲康唑II型晶体生长的抑制作用明显高于对其I型的作用。研究指出在晶体生长过程中,高分子聚合物会吸附到晶体表面,改变晶体-流体界面能,调控药物的晶体生长能垒,进而影响晶体生长[44]。Shi等[45]通过宽频介电技术测得高分子聚合物聚环氧乙烷添加前后无定形药物体系分子运动性的改变情况,发现整体分子运动的改变不足以解释高分子聚合物对晶体生长的影响,推测二元体系晶体生长极有可能是整体分子运动与界面高分子富集共同调控的。在随后研究中,Zhang等[46-47]通过偏光显微镜,拉曼成像及X-射线微区分析获取了高分子聚合物富集于药物晶体生长前沿界面的直接证据。更为重要的是,研究表明高分子聚合物在不同晶型晶体生长界面的富集情况存在显著差异,且该富集情况与高分子聚合物选择性影响药物不同晶型晶体生长速率直接相关(见图7)[47]。

图7 聚环氧乙烷在非甾体抗炎镇痛药吲哚美辛不同晶型结晶生长界面富集拉曼成像[47]
4 无定形药物溶出及表界面影响
无定形药物拥有更高的自由能,且其在溶出过程中无需打破晶格结构,研究表明其通常具有优于其晶体形式的溶出速率和更高的过饱和度,有利于获得更优的生物利用度[48]。为实现上述目标,在溶液中有效维持药物无定形态或减缓其结晶极为重要。目前有多种技术手段可用于维持药物的无定形或减缓无定形药物溶液结晶,包括以高分子聚合物为基质的无定形固体分散体[48],以小分子为配体的共无定形[49],以及以介孔材料为载体的介孔载药体系等技术[50]。如高分子聚合物与无定形药物组成的固体分散体,药物与高分子可形成特定的相互作用,一定程度上抑制或减缓药物结晶,从而有效延长其过饱和维持时间[51-52]。近年研究发现高分子聚合物可吸附于无定形及晶体药物表面,阻止溶液药物分子进一步参与结晶[53]。此外,高分子在固液界面上的分子构象也会显著影响其抑晶效果及过饱和维持能力。Schram等[54]研究发现醋酸羟丙甲纤维素琥珀酸酯(HPMCAS)在pH 3条件下,在固液界面呈未盘绕的球状,此时抑晶效果较差,当pH升高至6.8时,HPMCAS均匀分布于固液界面,有效减少晶体生长的可能位点,可有效抑制或减缓无定形药物结晶,并有效维持体系过饱和。
同样的,界面也是介孔载药体系药物溶出需要重点关注的内容。介孔载体表面积、孔道界面结构、表界面官能团组成等均是影响药物溶出的关键因素[55-56]。研究表明药物-介孔材料界面形成强相互作用时有利于药物缓释,相反,弱的药物-界面相互作用则有可能导致药物突释[50]。Ukmar等[57]研究发现吲哚美辛从介孔硅材料SBA-15及CMC-41的药物释放动力学与建立在药物-孔道界面相互作用基础上的预测值基本符合。他们指出药物-孔道界面可决定孔道结构中药物的局部流量及最大药物释放量,从而导致药物释放的孔道尺寸依赖性[57]。此外,研究发现药物吸附于介孔材料表面也是导致介孔载药体系药物非完全释放的重要原因之一。McCarthy等[58]研究调血脂药物非诺贝特从介孔硅材料中的释放,发现有70 %~80 %的药物在溶出开始的1 h内释放,但随时间延长药物释放量不再增加。他们指出药物在介孔材料表面吸附及小部分药物分子处在溶出介质无法润湿的孔道内部是发生上述现象的主要原因[58]。在该研究中,表面活性剂被证实可有效增加非诺贝特在介孔载药体系中的释放量,除增强溶出介质润湿性外,表面活性剂本身可与药物竞争吸附于界面材料表面,从而导致药物释放增加[58]。最新研究报道药物-介孔材料界面的相互作用主要依赖于形成氢键或者离子键相互作用[59-60]。Hate等[60]以酮康唑、氯氮平及阿扎那韦为模型药物研究药物-介孔硅材料相互作用对介孔载药体系药物溶出的影响。研究证实溶出介质pH升高可导致药物释放量减少,主要源于pH升高可显著增强介孔材料表面负电性,有效增加带正电荷药物的界面吸附[60]。
5 总结和展望
无定形药物制剂的物理稳定性和溶出是影响无定形药物制剂开发的关键问题,但其深层机制尚不明确。无定形药物的物理稳定性失稳和溶出通常最早都是发生在其表界面,且这些现象无法通过整体或体相性质进行合理解释。因此在研究中需深入研究表界面效应对无定形态药物成核、晶体生长,相分离及溶出等方面的影响,有助于更为客观、更符合实际应用地阐明无定形态药物的物理稳定性和溶出的深层次机制。此外,量化表界面效应引入的特殊相分离、成核及晶体生长行为等带来的对物理稳定性和溶出的影响对于未来预测模型的建立和优化尤为关键,直接关系到预测模型的科学合理性和准确性。从无定形药物制剂的过程质量控制角度出发,无定形药物制剂从处方前研究到最终产品问世需经历多个关键的技术环节,在众多技术环节中难以避免地会引入新的界面或改变原有表面,如何有效避免和调控表界面也是关键问题之一。因此,伴随着对表界面效应的进一步深入研究和挖掘,无定形药物制剂的物理稳定性和溶出会有更深层次的机制解释,从而有利于在未来开发更多质量稳定可控的市售制剂。
猜你喜欢
青岛科技大学学报(自然科学版)(2023年6期)2023-11-25 17:17:56
上海理工大学学报(2021年3期)2021-07-20 08:04:10
人工晶体学报(2021年6期)2021-07-12 07:59:58
陶瓷学报(2020年2期)2020-10-27 02:16:14
中国材料进展(2020年4期)2020-05-23 02:15:52
中国材料进展(2019年10期)2019-12-07 05:32:54
人工晶体学报(2019年8期)2019-09-18 02:53:02
火工品(2019年3期)2019-09-02 05:48:00
西北药学杂志(2018年5期)2018-09-20 11:27:00
无机盐工业(2017年6期)2017-03-11 14:43:47
