
HPLC 法测定32%苯嘧磺草胺·草甘膦可分散油悬浮剂中甲醛的含量
2022-12-08 08:36:44韦沙迪张小敏李玮松吴丽文王爱臣
世界农药 2022年11期
韦沙迪,张小敏,李玮松,吴丽文,王爱臣
(惠州市银农科技股份有限公司,广东 惠州 516257)
苯嘧磺草胺(saflufenacil),化学名称N-[2-氯-4-氟-5-(3-甲基-2,6-二氧-4-(三氟甲基)-3,6-二氢-1(2H)-嘧啶)苯甲酰]-N-异丙基-N-甲基硫酰胺,是一种新的原卟啉原氧化酶抑制剂类除草剂。它对杂草生长的抑制效应是通过对光合系统的抑制而触发的,可在光下迅速诱导植物组织坏死,降低同化作用。该剂的作用机制为抑制四吡咯生物合成途径中的原卟啉原IX氧化酶(PPO)活性,造成原卟啉IX(Proto)及过氧化氢在叶片组织中积累,在光下,细胞液中Proto分子与氧反应形成单态氧与氧基,从而使细胞膜的不饱和脂肪酸过氧化,造成膜的透性与功能迅速丧失,叶绿体色素白化,组织坏死,最终生长受抑制直至植株死亡[1]。苯嘧磺草胺是广谱性触杀型除草剂品种,可防除70种以上阔叶杂草,包括多种耐草甘膦杂草,如藜、豚草、问荆、猪毛菜、水麻以及苘麻、苍耳、反枝苋、播娘蒿、离子芥、地肤、卷茎蓼等大多数阔叶杂草及部分禾本科杂草。
草甘膦(glyphosate),化学名称N-磷羧基甲基甘氨酸,属有机磷类内吸传导型除草剂,具有广谱灭生性,主要通过抑制植物体内5-烯醇丙酮酰莽草酸-3-磷酸合成酶,从而抑制莽草素向苯丙氨酸、酪氨酸及色氨酸的转化,使蛋白质的合成受到干扰,导致植物死亡[2]。草甘膦杀草谱广,对40多科100多种杂草有较好的防除作用,包括单子叶和双子叶、一年生和多年生、草本和灌木等植物。豆科和百合科的部分植物对草甘膦的抗性强。草甘膦进入土壤后很快与铁、铝等金属离子结合而失去活性。
32%苯嘧磺草胺·草甘膦可分散油悬浮剂是由草甘膦与苯嘧磺草胺2种作用机制不同的药剂混配而成的茎叶处理除草剂,2者复配,防治谱更为全面,防效增强,同时可延缓杂草抗药性,并有效治理生产上对草甘膦产生抗性的杂草。
甲醛作为草甘膦的合成原料,在草甘膦原药及相关制剂中有残留,因为甲醛具有致癌和致突变,甲醛含量的控制变得尤为重要。目前草甘膦中甲醛的测定方法主要依据国标《GB/T 12686—2017 草甘膦原药》,标准中采用紫外分光光度计法进行甲醛含量的测定,但由于可分散油悬浮剂基质比较复杂,滤出的样品难以完全澄清透明,导致测定结果偏高,难以实现精准定量。32%苯嘧磺草胺·草甘膦可分散油悬浮剂中甲醛的限度要求≤0.8 g/kg,本文采用高效液相色谱法,通过Hantzsch试剂与甲醛反应生成DDL衍生物,测定32%苯嘧磺草胺·草甘膦可分散油悬浮剂中甲醛的含量。反应方程式如下:
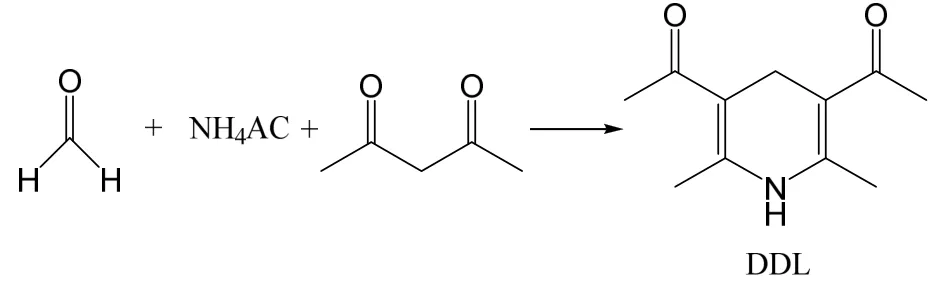
该分析方法具有操作简便、无样品基质干扰、分离效果好等特点,准确度和精密度均能达到定量分析要求,可作为企业生产过程中该产品质量控制方法。
1 试验部分
1.1 仪器
Waters e2695 型高效液相色谱仪配2489 型紫外检测器及Empower 3 色谱工作站和自动进样器(沃特世科技有限公司);JP040S 超声波清洗机(深圳洁盟超声波设备有限公司);XSE205DU 型分析天平(0.01 mg,梅特勒托利多);Waters XBridge C18色谱柱(5 μm,250 mm×4.6 mm 不锈钢柱,沃特世科技有限公司)。
1.2 试剂
甲醛标样(质量分数为37.1%,Fisher);乙腈(色谱纯,Merck);水:新蒸二次蒸馏水;乙酸铵(分析纯,国药集团化学试剂有限公司);乙酸(分析纯,天津市科密欧化学试剂有限公司);乙酰丙酮(分析纯,国药集团化学试剂有限公司)。
1.3 液相色谱操作条件
以乙腈与水作为流动相,2 者比例为25∶75(体积比);流速:1.0 mL/min;柱温:30 ℃;检测波长:412 nm;进样体积:5µL。在上述色谱条件下,甲醛的衍生物DDL 保留时间约为5.4 min。上述操作参数是典型的,可根据不同仪器特点,对给定的操作参数作适当调整,以期获得最佳效果。典型的标样和试样高效液相色谱图分别见图1、2。
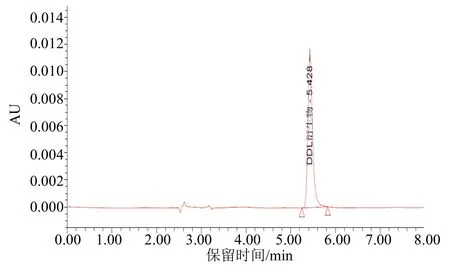
图1 甲醛标样衍生物DDL 高效液相色谱图
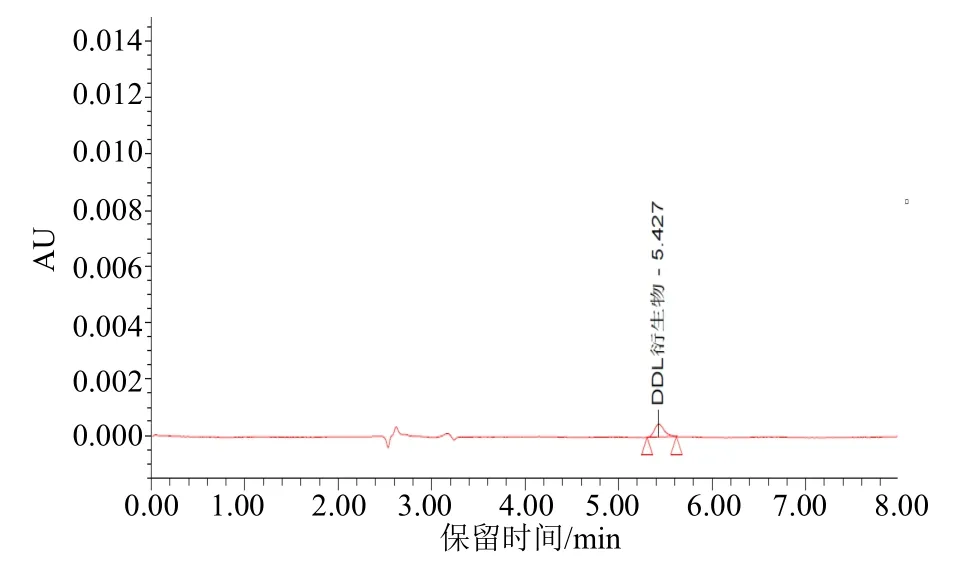
图2 32%苯嘧磺草胺·草甘膦可分散油悬浮剂试样中甲醛衍生物DDL 高效液相色谱图
1.4 测定步骤
1.4.1 Hantzsch 试剂的配制
称取15 g乙酸铵置于100 mL的容量瓶中,加入0.3 mL乙酸和0.2 mL的乙酰丙酮,加入适量的水超声使溶解后,冷却,加水定容,摇匀。
1.4.2 标样溶液的配制
称取0.27 g甲醛标样(精确至0.000 2 g),置于1 000 mL容量瓶中,加入适量水,超声溶解后冷却,加水定容;再移取1 mL该溶液,置于100 mL容量瓶中,加水定容,摇匀。取5 mL标样溶液和5 mL Hantzsch试剂等比例混合,摇匀,在室温下放置至少2 h,过滤,作为标样溶液。
1.4.3 试样溶液的配制
称取0.33 g左右的试样,置于100 mL容量瓶中,用水稀释至刻度,加盖后超声10 min使试样溶解,冷却,摇匀。取5 mL试样溶液和5 mL Hantzsch试剂混合,摇匀,室温放置至少2 h,过滤,作为试样溶液。
1.4.4 测定
在上述操作条件下,待仪器基线稳定后连续注入数针标样溶液,直至相邻2针的相对响应值变化小于1.5%,即可按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行测定,记录峰面积。
1.4.5 计算
将测得的2针试样溶液及试样前后2针标样溶液中的甲醛衍生物DDL峰面积分别进行平均,试样中甲醛的质量分数X(g/kg)按下式计算:
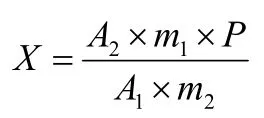
式中:A1为2针标样溶液中甲醛衍生物DDL峰面积的平均值;A2为2针试样溶液中甲醛衍生物DDL峰面积的平均值;m1为甲醛标样的质量,g;m2为试样的质量,g;P为标样中甲醛的质量分数,%。
1.4.6 允许差
甲醛2次平行测定结果之相对差应不大于10%,取其算术平均值为测定结果。
2 结果与讨论
2.1 色谱条件的选择
通过岛津LC-20AT高效液相色谱仪光谱数据采集功能,获得甲醛衍生物DDL紫外波长扫描图。该物质最大吸收波长为412 nm,所以将检测波长定为412 nm。色谱柱选择常用的反相填料Waters XBridge C18柱。流动相选择乙腈+水,对二者不同比例进行试验发现当乙腈和水的体积比为25∶75时,色谱峰峰形对称,基线平稳,分析时间较短,利于日常生产监控。
2.2 分析方法的线性关系试验
称取甲醛标样0.273 9 g,置于1 000 mL容量瓶中,加入适量水,超声溶解后冷却,加水定容作为母液;移取上述标样母液0.01、0.05、0.1、0.5、1、1.5 mL分别于不同的100 mL容量瓶中,用水定容,摇匀,作为线性溶液储备液。分别取5 mL线性溶液储备液和5 mL Hantzsch试剂混合,摇匀,室温放置至少2 h,过滤,作为测定的线性溶液。甲醛衍生物DDL线性关系方程y=0.071 2x+2 666.1,线性相关系数为0.997(图3)。
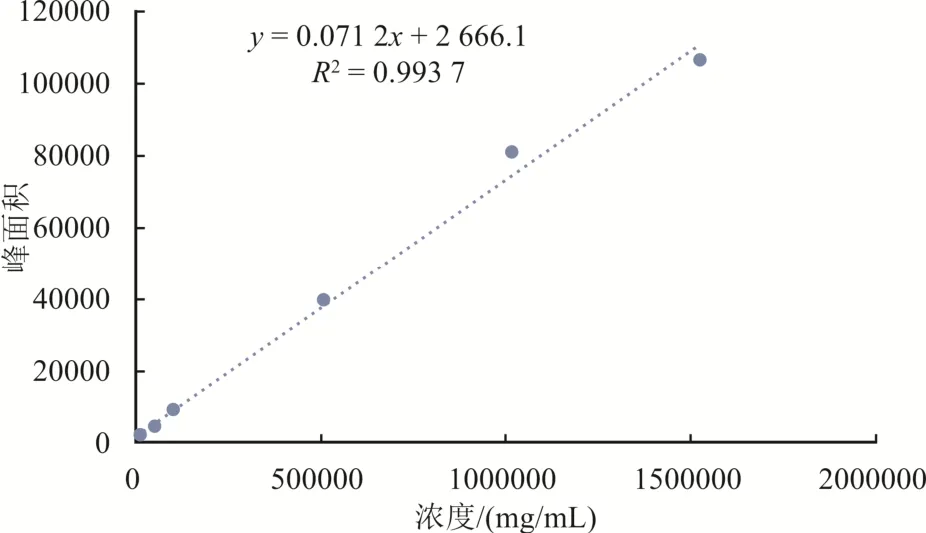
图3 甲醛衍生物DDL 线性关系图
2.3 分析方法的精密度试验
准确称取5份32%苯嘧磺草胺·草甘膦可分散油悬浮剂试样,在上述色谱操作条件下进行分析,相对标准偏差(RSD)为5.9%(表1)。
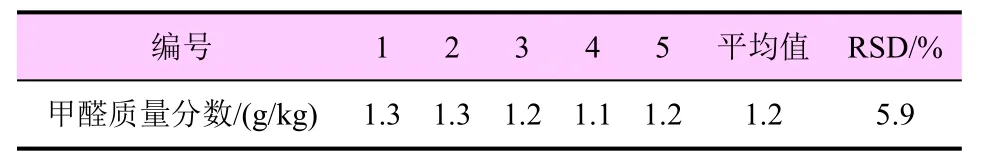
表1 分析方法的精密度试验结果
2.4 分析方法的准确度试验
称取0.27 g甲醛标样,置于1 000 mL容量瓶中,加入适量水,超声溶解后冷却,加水定容作为母液。参照NY/T 2887—2016农药产品质量分析方法确认指南中准确度的测定方法,取样量减半(样品称样量为0.15 g),称取约0.15 g的试样置于100 mL量瓶中,按1∶1比例加入标准品,即移取0.5 mL标样母液至于100 mL容量瓶中,加水定容,摇匀;取5 mL标样溶液和5 mL Hantzsch试剂等比例混合,摇匀,在室温下放置至少2 h,过滤。按照上述方法共制成5个样品,分别对每个样品中的甲醛含量进行测定。甲醛平均回收率为105.8%,该方法回收率良好,测定结果准确可靠(表2)。
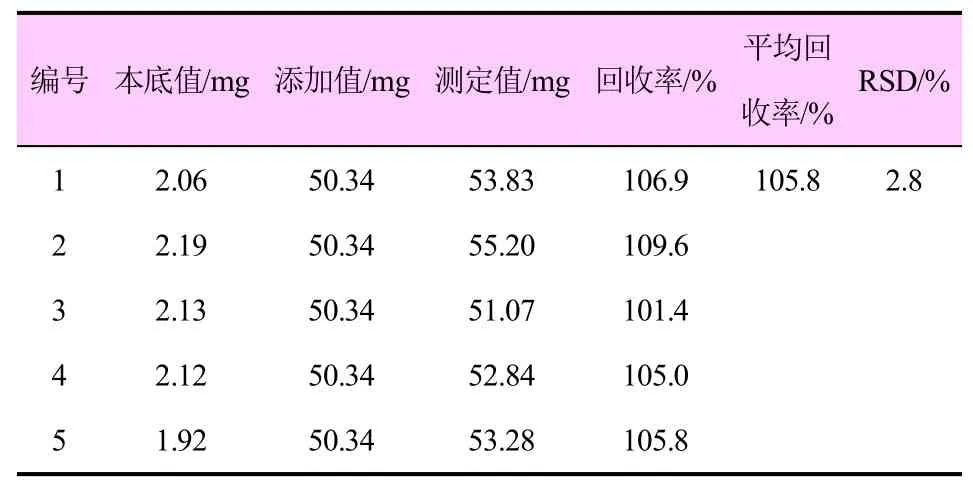
表2 分析方法的准确度试验数据
2.5 分析方法的定量限和检出限
称取甲醛标样0.273 9 g,置于1 000 mL容量瓶中,加入适量水,超声溶解后冷却,加水定容作为母液;移取上述标样母液0.01 mL置于100 mL容量瓶中,用水定容,摇匀,制成浓度为10 161.7 mg/mL的溶液,再移取该溶液5 mL至10 mL的容量瓶中,得到定量限溶液储备液。分别取5 mL该储备溶液和5 mL Hantzsch试剂混合,摇匀,室温放置至少2 h,过滤,即得信噪比约为10∶1的定量限溶液。该分析方法的定量限为0.003 g/kg。该分析方法的检出限为0.0015 g/kg。

图4 甲醛衍生物DDL 定量限高效液相色谱图
3 结 论
试验结果表明甲醛质量浓度为0.010~1.520 mg/L时(r=0.997),该方法线性关系良好;该方法精密度相对标准偏差为5.9%;平均回收率为105.8%。该分析方法的定量限为0.003 0 g/kg,检出限为0.001 5 g/kg。该方法的准确度和精密度较高,线性关系良好,具有简便、快速、准确及分离效果好的优点,是一种可行的分析方法。值得注意的是如果试剂本身含有甲醛,那么Hantzsch试剂本身会带来较高的背景吸收,此时,需同时进行空白溶剂的分析。
猜你喜欢
疯狂英语·读写版(2024年1期)2024-03-13 13:22:55
理化检验-化学分册(2022年11期)2022-11-27 05:21:26
化工管理(2021年4期)2021-02-27 07:34:08
武夷科学(2019年2期)2019-12-20 08:39:14
农药科学与管理(2019年6期)2019-11-23 08:17:10
环境科技(2015年4期)2015-11-08 11:10:48
绿色科技(2015年2期)2015-03-27 04:29:56
营销界(2015年23期)2015-02-28 22:06:12
化工科技(2014年5期)2014-06-09 06:11:24
山东农药信息(2013年10期)2013-11-27 02:16:02
