
Generation of sulfonylureas under photoredox catalysis and their biological evaluations
2022-12-07 08:26XuefengWngJunZhngQiChenWeiZhouJieWu
Chinese Chemical Letters 2022年11期
Xuefeng Wng, Jun Zhng, Qi Chen,∗, Wei Zhou, Jie Wu,c,d,∗
a Taizhou Central Hospital (Taizhou University Hospital) & School of Pharmaceutical and Materials Engineering, Taizhou University, Taizhou 318000, China
b Department of Chemistry, Fudan University, Shanghai 200438, China
c State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
d School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
Keywords:Sulfonylurea Chlorosulfonyl isocyanate Photocatalysis
ABSTRACT Traditional synthesis of sulfonylureas largely depends on nucleophilic addition of arylsulfonamides to presynthesized isocyanates.Now we report a new access to alkylsulfonylureas with good yields and broad substrate scope.With the insertion of commercialized chlorosulfonyl isocyanate under photoredox catalysis, alkylsulfonylureas are synthesized in one-pot from the corresponding anilines and silyl enolates.A reaction mechanism is proposed showing the transformation undergoes a radical process, and the practicality of this methodology is proven via application to bioactive molecules.Additionally, the anti-cancer and anti-virus screening of these compounds is evaluated.
Due to their unique physiological activities, sulfonylureas are widely studied in medicinal and pharmaceutical chemistry [1–4].With their abilities to combine with receptors on cell plasma membranes [5–7], a series of sulfonylurea-based anti-diabetic drugs and hypoglycemic agents were developed in the 20thcentury, together with the applications in antibiotics and herbicides(Scheme 1) [8–15].Because of their distinctive activity and enormous practical application value, synthesis of sulfonylureas has aroused great research interest of many scientists.
Traditional synthesis of sulfonylureas largely depends on the addition of sulfonamides to isocyanates (Scheme 2a) [16,17].In cases that sulfonyl isocyanates are accessiable (R1=Ph,p-Tol,etc.),addition of primary amines to sulfonyl isocyanates is also applied(Scheme 2b) [18,19].In some other situations, substitutions of carbonamides by primary sulfonamides could give rise to the desired sulfonylureas as well (Scheme 2c) [20–22].However, all these synthetic routes are largely limited to the use of pre-synthesized isocyanates and carbonamides, introducing problems of functional group tolerance and step efficiency.Moreover, R groups of easyaccessed sulfonamides and sulfonyl isocyanates are mostly aromatic or benzylic on the whole, and a single-step synthesis of alkylsulfonylureas has not yet been achieved.Therefore, it is imperative and promising to establish a single-step synthetic methodology for sulfonylureas.
In order to provide a one-step method for the synthesis of alkylsulfonylureas, a couple of reaction pathways were designed and explored by our group.Restricted to the weak nucleophilicity of amino group in urea molecules, it could hardly coordinate with the catalytic center in the transmetallation step during a typical metal-catalyzed process with the insertion of sulfur dioxide(Scheme 3a) [23–31].Additionally, attempts of tuning the electrical nature of urea molecule by chlorination also didn’t bring positive results (Scheme 3b).
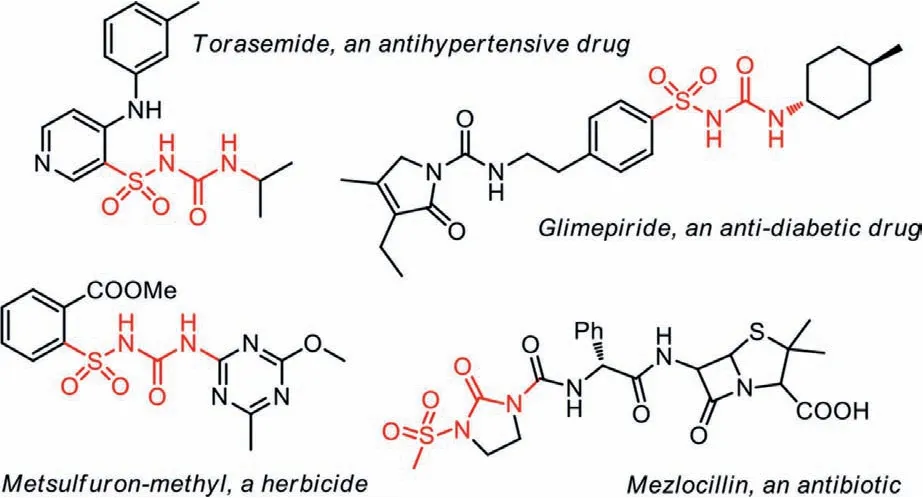
Scheme 1 .Several examples of bioactive sulfonylureas.
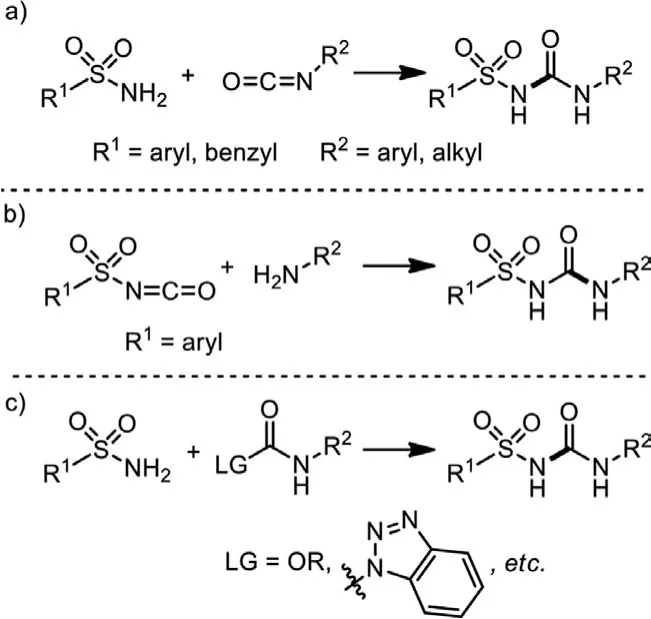
Scheme 2 .Traditional access to sulfonylureas.
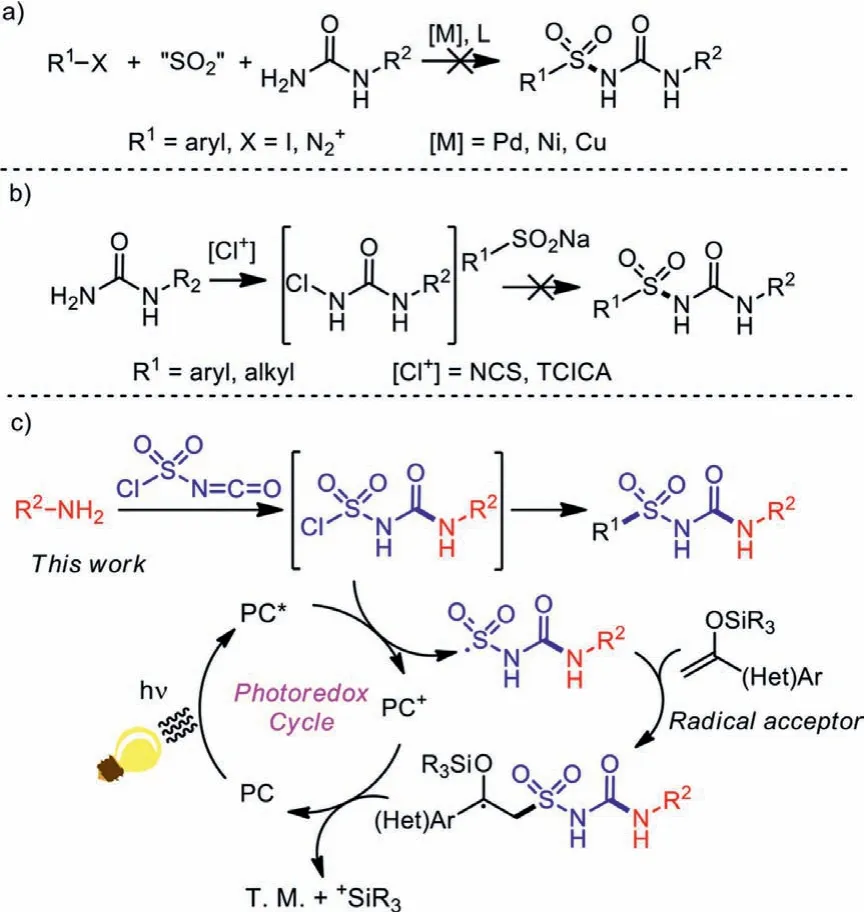
Scheme 3 .Design of the reaction pathway.
After encountering obstacles in experiments with urea-based raw materials, we then tried to generate the urea fragment during the coupling reaction, and chlorosulfonyl isocyanate (CSI), a widely-studied reagent in the 20thcentury, aroused our interest.Chlorosulfonyl isocyanate is a commercially-available, cheap and low-toxic raw material, which is widely used in organic and drug synthesis [32–36].With both sulfonyl chloride and isocyanate functional groups, CSI often serve as a dielectrophile in organic synthesis, giving rise to functionalized aminosulfonylureas.In addition to coupling to two nucleophiles, other transformations of CSI were also studied in the past few decades.After combination with amines, intramolecularortho-Friedel-Crafts sulfonylation would occur under Lewis-acid catalysis to produce a range of 1,2,4-benzothiazidazin-3(4H)-one 1,1-dioxides [37], while intermolecular sulfonylation of indole derivatives could give indole-3-sulfonylureas, a series of acetolactate synthase inhibitors [38].The electrophilic isocyanate part of CSI was reported to keep inert during coupling with arylstannates under mild conditions, giving arylsulfonyl isocyanates as products [39,40].However, coupling routes other than aromatic electrophilic substitutions are rarely studied.
With understanding of the reactive nature of CSI, we tried to adapt CSI to double coupling reactions, with an expectation to synthesize sulfonylureas in a single step.Due to the elimination ofβhydride, transition metal catalysis is not suitable for further transformations, and a radical process was introduced (Scheme 3c).According to our expectations, an amine substrate should first re-act with the more-electrophilic isocyanate side of CSI, leading to a chlorosulfonyl urea intermediate.Then the survived sulfonyl chloride side could be reduced to sulfonyl radical under photoredox catalysis through single electron transfer (SET), which would be captured by a radical acceptor R1reagent to afford an alkylsulfonylurea product.
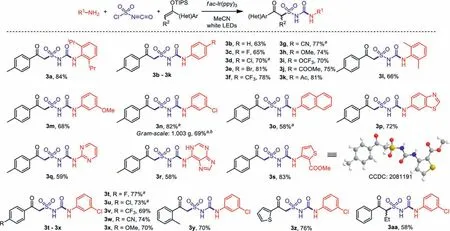
Scheme 4 .Substrate scope for the photoinduced synthesis of alkylsulfonylureas.Conditons: 1 (0.2 mmol), chlorosulfonyl isocyanate (0.21 mmol, 1.05 equiv.), 2 (0.5 mmol,2.5 equiv.), fac-Ir(ppy)3 (2.0 mol%), CH3CN (3.0 mL), 80 W white LEDs, 16 h.Isolated yield based on 1. a Yields after filtration and recrystallization. b Reaction time was 40 h.
In order to provide a general direction for the desired process, a chlorosulfonyl urea A was synthesized according to reported methods [41], and a highly-reactive enol ether 2a was selected as a radical acceptor.In our first attempts, the desired product 3a was obtained in 48% yield in the presence offac-Ir(ppy)3with potassium metabisulfite (K2S2O5) as base in MeCN (Table 1, entry 1),while other photocatalysts gave inferior results (Table 1, entries 2-7).Surprisingly, the yield was found significantly increased to 85%when the base was omitted, showing that the reaction didn‘t require participation of additional bases or reductants (Table 1, entry 8).Then during our further investigations focusing on one-pot synthesis from 2,6-diisopropylaniline 1a, it was found that a more powerful 80 W white LED lamp could further improve the transformation (Table 2, entries 1-3), while other solvents didn’t provide better results (Table 2, entries 4-7).Moreover, it was discovered that the hygroscopic CSI should be handled under Schlenk techniques.
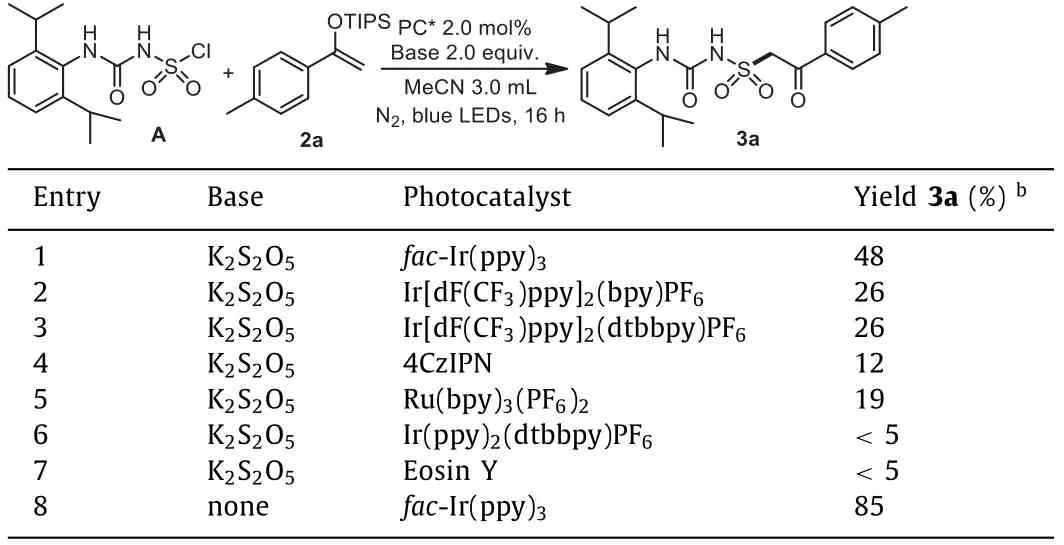
Table 1 Initial investigations using chlorosulfonyl urea A.a
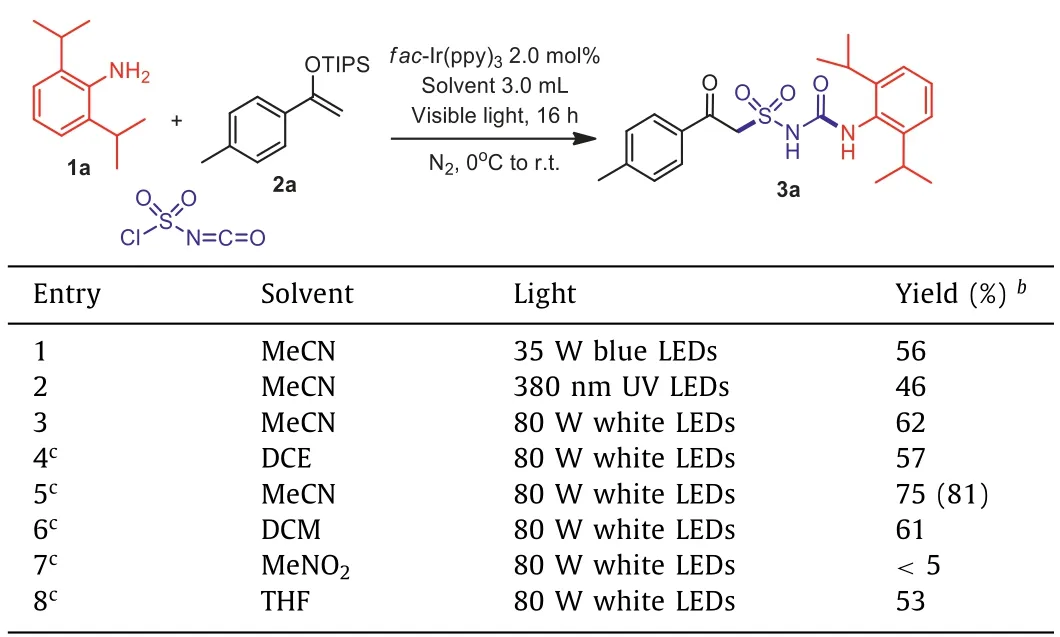
Table 2 Optimization of one-pot reaction conditions. a
With the established “standard conditions” in hand, the substrate scope of this three-component transformation was then studied (Scheme 4, for detailed procedure, see Supporting information).With various substituted anilines, the desired products 3a–3p were obtained in good yields.In some case as 3d, 3l and 3n,the formed precipitate was directly filtered from the reaction mixture, then washed and recrystallized to give analytically pure products.A gram-scale reaction was also carried out with 4.0 mmol of 3-chloroaniline 1n, and 1.003 g of product 3n was obtained smoothly after workup.Heterocyclic amines also worked well, as products with pyrimidine (3q), adenine (3r) and thiophene (3s)were achieved in good yields.X-ray crystal structure of compound 3s was measured and confirmed (CCDC: 2081191).With respect to the enol ethers, it was found that reactions of both electronrich and electron-deficient substrates provided good results, whileortho-substituted product 3y and a thienyl product 3z were also obtained successfully.A phenylbutanone-derived secondary enol ether was workable as well leading to the desired product 3aa in 58% yield.However, alkyl enol silyl ethers were not applicable in this transformation.For instance, the reactions of acetone and cyclohexanone-derived enol silyl ethers could not give the desired products.
After confirming the universality of the substrates, we then explored the late-stage functionalization of some bioactive molecules and market drugs.We were delighted to found that the amino groups in the molecules were smoothly transformed to the corresponding alkylsulfonylurea products (Scheme 5).Complex heterocycles as the imidazo[b]quinoline inImiquimodwas successfully converted to the functionalized product 3ac, while the amide groups inLenalidomideandSulfadiazinewas also tolerated (compounds 3ad and 3ae).
In addition to anilines and heterocyclic amines, we also tried to expanded the substrate scope to alkyl and secondary amines.AsNmethyl-1-phenylmethanamine 1ag was employed as the synthon,product 3ag was obtained in 37% yield as our expectation, showing that this method could also be applicable to secondary amines or alkyl amines.With fluorine substitutions on enol ether 2ah,the transformation also resulted in the desired difluoro-substituted product 3ah in moderate yields (Scheme 6).
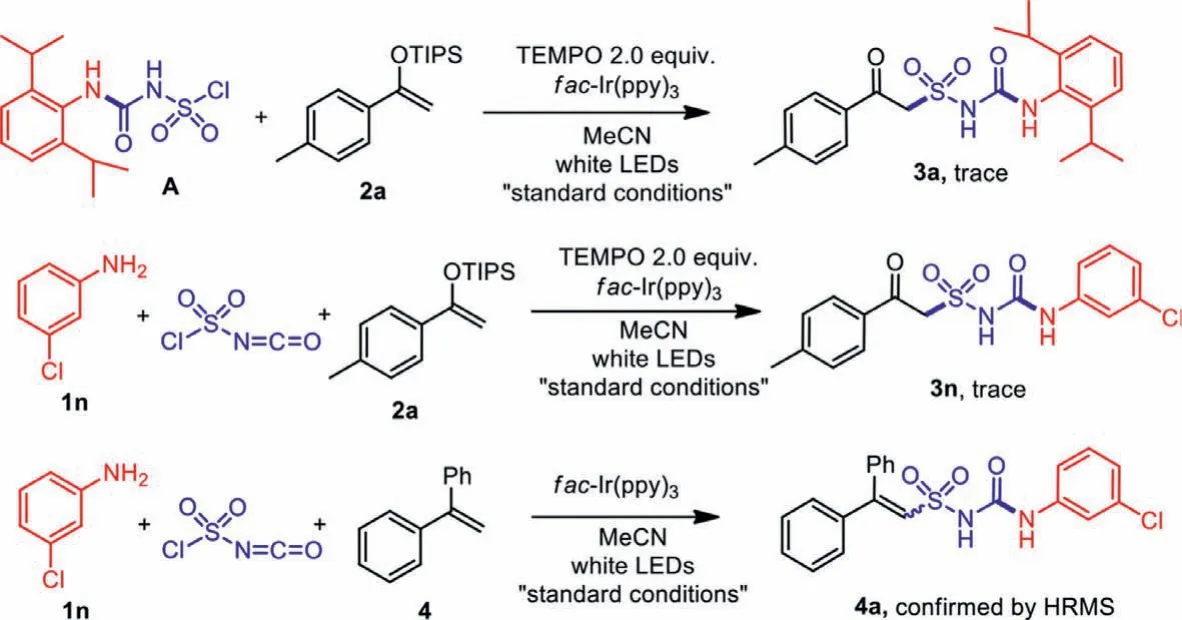
In order to confirm the reaction mechanism, radical inhibitor TEMPO (2.0 equiv.) was added to the reaction of pre-synthesized intermediate A and enol ether 2a, together with the threecomponent reaction of 3-chloroaniline 1n, chlorosulfonyl isocyanate and enol ether 2a under “standard conditions”.Only trace amounts of product 3a or 3n was detected, proving the reaction to proceed in a radical pathway.When 1,1-diphenylethene 4 was occupied as the radical acceptor in the place of enol ether 2, a Heck-type deprotonated product 4a was produced, which further verified the proposed mechanism (Scheme 7).
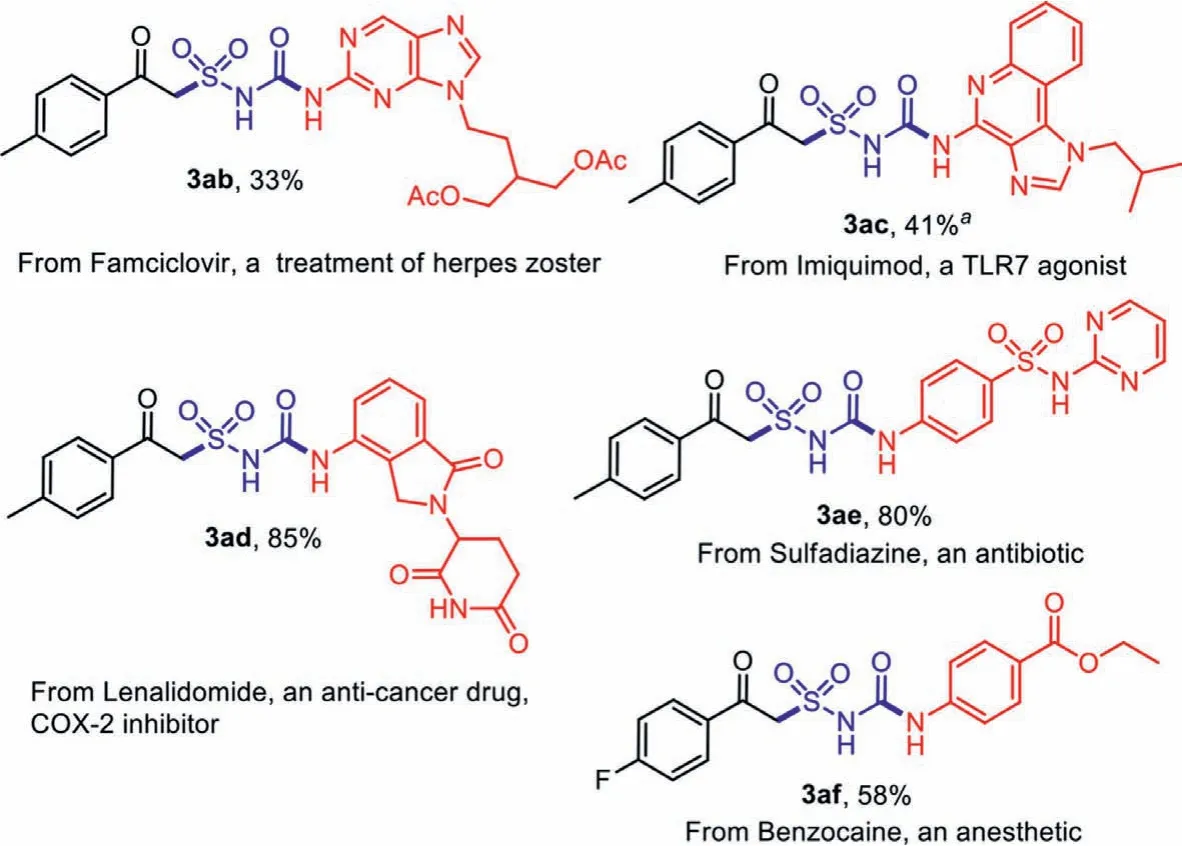
Scheme 5 .Late-stage functionalization of bioactive molecules.Isolated yield based on amines 1. a Yields after filtration and recrystallization.
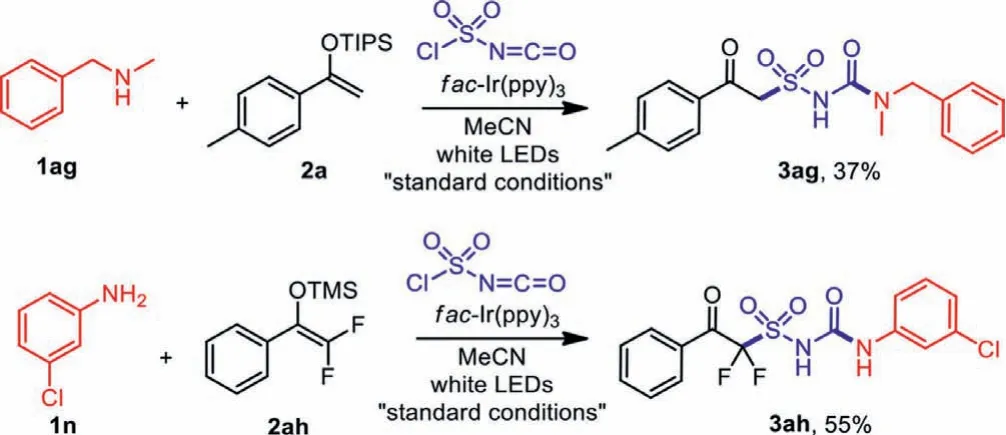
Scheme 6 .Additional transformations.
Scheme 7 .Mechanism investigations.
Next, some of these novel alkylsulfonylurea derivatives were applied to anti-cancer and anti-virus screening in order to find potential lead structures for possible drug discovery.First, the cytotoxicity of selected compounds on four human cancer cell lines, including T24 (bladder cancer cell), PC-3 (prostate cancer cell), HO1809(ovarian cancer cell), and SKOV3 (ovarian cancer cell), was evaluated using MTT assay.It was found that, beside compound 3ac, all cell lines still maintained ≥90% viability for the tested compounds at the concentrations of 25 and 50 μmol/L (Table 3), indicating the low cytotoxicity of these new alkylsulfonylureas.For compound 3ac, it showed a concentration dependence cytotoxicity after 48 h of incubation and the cell proliferation inhibition reached about 30% at 50 μmol/L.Although the activity is relatively weak, it supplies a promising structure skeleton for further optimization.
In anti-virus activity assay, Vero cells were infected with 1000×the median tissue culture infectious dose (1000×TCID50) of VSV (Vesicular stomatitis virus), RSV (Respiratory syncytial virus),and HSV-1 (Type 1 herpes simplex virus), and MDCK cells were infected with 10×TCID50of H3N2 (Influenza A virus), and then the cells were transferred to the tested compound solutions (diluted in serum-free F12/DMEM medium) for 72 h.Ribavirin was used as positive control.The results showed that only compound 3ac could inhibit influenza-induced cell apoptosis effectively, with about 50%increase in cell viability (Table 4), while others had no activity at all to any virus.To further confirm the inhibitory activity against H3N2 of 3ac, a series of concentrations were evaluated (Table 5).A concentration dependent anti-influenza activity was definitely observed.Besides, more meaningful, 3ac showed a better potency in CPE (cytopathic effect) inhibition than Ribavirin (Fig.1).Thus,compound 3ac might be a good lead compound for anti-influenza drug development, even it had a lower cell viability than Ribavirin which might induced by its cytotoxicity what needed to be overcome in the further structure optimization.
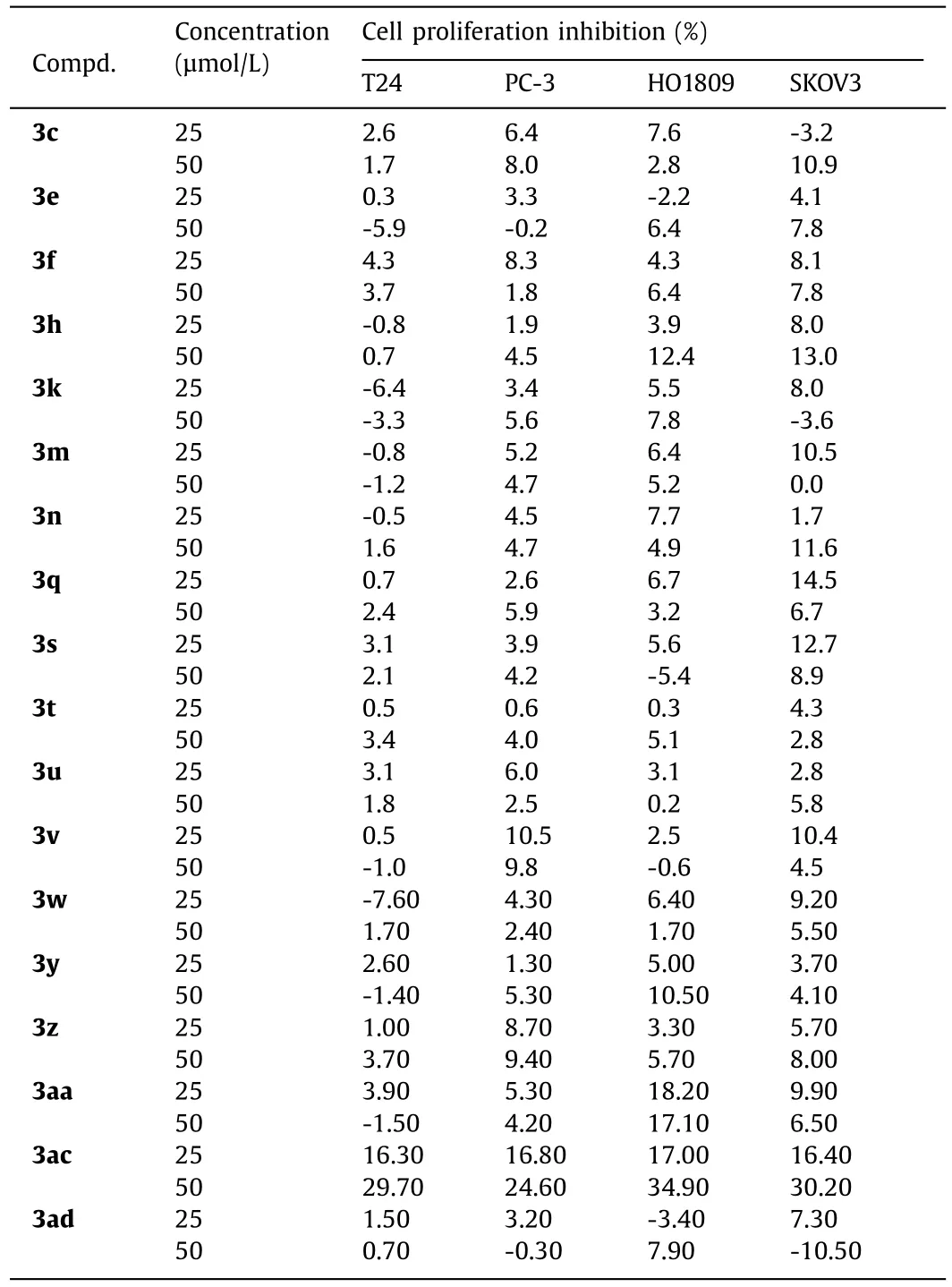
Table 3 Cytotoxicity of tested compounds on different cancer cells.
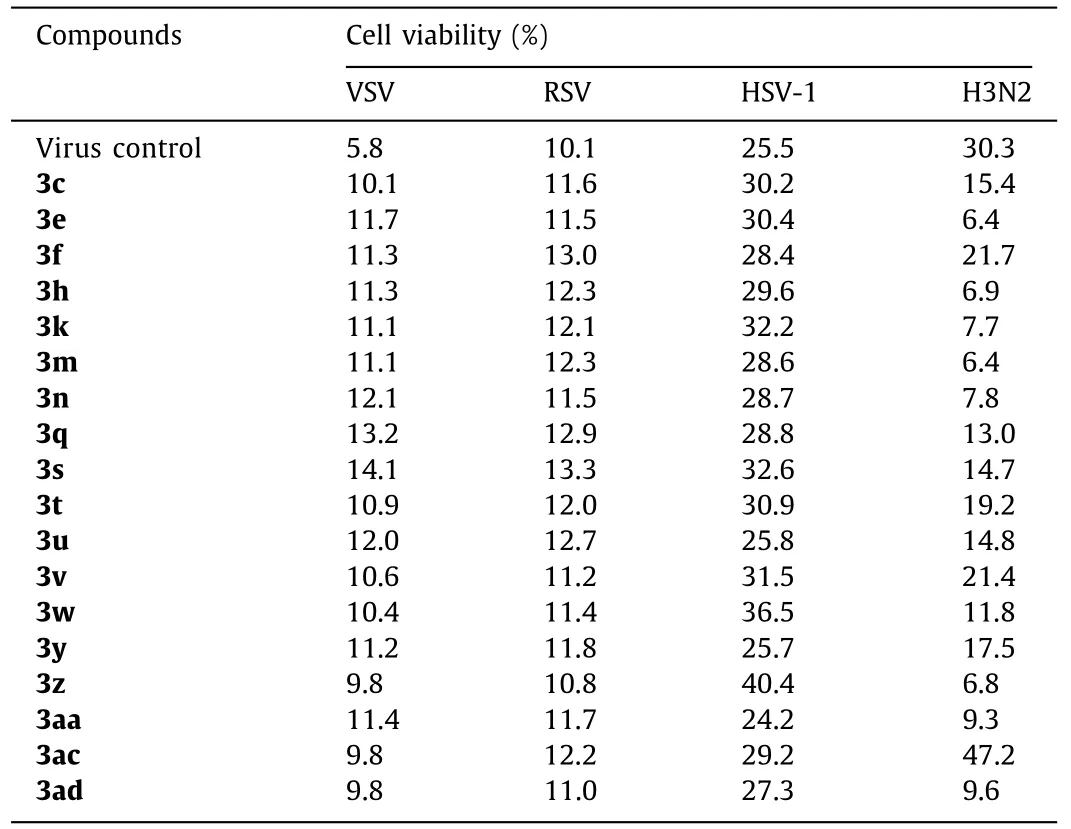
Table 4 In vitro antiviral effects against four kinds of viruses in concentration of 50 μmol/L.

Table 5 Antiviral effect of 3ac against H3N2 influenza virus.
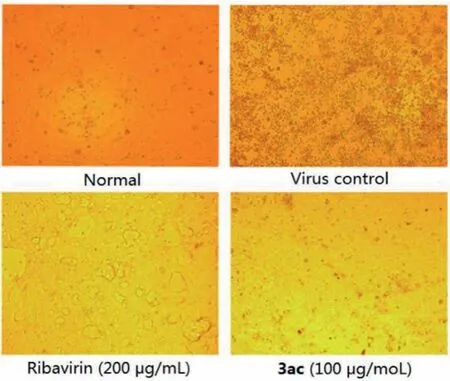
Fig.1 .CPE images of MDCK cells (Magnification×200).
In summary, we established a new method to access alkyl sulfonylureas from the corresponding amines, chlorosulfonyl isocyanate and alkyl nucleophiles.With the formal “insertion” of CSI, the desired sulfonylureas were formedviaain-situgenerated chlorosulfonyl urea intermediate.Various functional groups are tolerated, and the transformation was confirmed to be suitable for late-stage functionalization of market drugs and bioactive molecules.A radical reaction mechanism was clearly proposed, revealing the path of single electron transfer (SET) in the reaction process.Furthermore, compound 3ac is found to have a remarkable anti-influenza virus efficacy as a lead compound or a potential alternative.With extensively-available amines and cheap, commercially available chlorosulfonyl isocyanate, it is anticipated that a number of new sulfonylurea molecules could be achieved with this method.We look forward to the adaption of this method in drug development and synthesis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Financial support from National Natural Science Foundation of China (No.21871053), the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No.2019R01005) and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (No.2020ZD04) is gratefully acknowledged.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.025.
 Chinese Chemical Letters2022年11期
Chinese Chemical Letters2022年11期
- Chinese Chemical Letters的其它文章
- Zeolite-based Fenton-like catalysis for pollutant removal and reclamation from wastewater
- 1,n-Thiosulfonylation using thiosulfonates as dual functional reagents
- Degradation of florfenicol in a flow-through electro-Fenton system enhanced by wood-derived block carbon (WBC) cathode
- Simultaneous determination of indole metabolites of tryptophan in rat feces by chemical labeling assisted liquid chromatography-tandem mass spectrometry
- Self-powered anti-interference photoelectrochemical immunosensor based on Au/ZIS/CIS heterojunction photocathode with zwitterionic peptide anchoring
- The role of Cs dopants for improved activation of molecular oxygen and degradation of tetracycline over carbon nitride