
1,n-Thiosulfonylation using thiosulfonates as dual functional reagents
2022-12-07 08:26DnhuGeJiWeiChenPeiXuJinyinPnXueQingChu
Chinese Chemical Letters 2022年11期
Dnhu Ge, Ji-Wei Chen, Pei Xu, Jinyin Pn, Xue-Qing Chu,∗
a Institute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, China
b Jiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, School of Pharmacy, Xuzhou Medical University, Xuzhou 221004, China
cSartorius Stedim (Shanghai) Trading Co., Ltd., Shanghai 200000, China
Keywords:Thiosulfonylation Thiosulfonates Dual functional reagents Difunctionalization Atomic economy
ABSTRACT In recent years, the direct introduction of sulfonyl and sulfenyl groups into unsaturated substrates by using thiosulfonates as unique dual functional reagents has inarguably provided chemists a new platform for the diverse synthesis of important S-containing derivatives.These 1,n-thiosulfonylation reactions usually feature simple procedures, 100% atom economy, and high regioselectivity.This review focuses on the recent advancements in the transformations of thiosulfonates through 1,n-thiosulfonylation involving the formation of two distinct C-S bonds under transition-metal-catalyzed or metal-free conditions, where thiosulfonates act as both a sulfonyl and a sulfenyl component.
1.Introduction
Organosulfur compounds not only widely appear in natural products [1], bioactive molecules, pharmaceuticals, and catalysts/ligands [2] but also are frequently used as versatile intermediates to prepare structurally diverse chemicals [3].For example,Brilinta (Ticagrelor) prevents platelets in our blood from sticking together; Trintellix (Vortioxetine) is an atypical antipsychotic and antidepressant for the treatment of the major depressive disorder (MDD); Otezla (Apremilast) is a phosphodiesterase 4(PDE4) inhibitor used to treat inflammatory autoimmune diseases;Tinidazole is a well-sold nitroimidazole antitrichomonal agent(Scheme 1A).Meanwhile, benefiting from the multivariate characteristics, the sulfenyl (-S-) and sulfonyl (-SO2-) functionalities can be easily converted to carbon-carbon single and double bonds[4,5].As a result, attractive advantages of sulfenyl and sulfonyl moieties have aroused great synthetic interests in developing efficient methodologies for incorporating these units together into one parent molecule.
Generally, transition-metal-catalyzed or photocatalyzed C-S bond formation reaction represents one of the most powerful ways to access organosulfur compounds during the past decades [5–10].In this regard, a diversity of sulfur-containing reactants including thiols [11], disulfides [12], sulfonyl hydrazides [13], sulfonates[14],etc.[15], have the capability to react as unique sulfur precursors for realizing sulfenylation and sulfonylation.Among them,thiosulfonates (S-esters of thiosulfonic acid) have been broadly harnessed as electrophilic sulfenylating sources, and the sulfonyl part is in comparison discarded as a waste (Scheme 1B, a) [16,17].Alternatively, they also can act as sulfonylating reactants by adjusting the inherent reactivity of the substrate (Scheme 1B, b) [18].Moreover, sulfonyl and sulfenyl radicals could be released through homolytically cleaving the S-SO2bond in thiosulfonates under metal-catalyzed or thermodynamic conditions [3].On the other hand, the regioselective and chemoselective difunctionalization of unsaturated substrates has become a central focus for the efficient synthesis of valuable molecules, especially starting from readily available building blocks [19–21].Recently, 1,n-thiosulfonylation reaction involving the installation of a sulfenyl group concomitant with the construction of C-SO2bond has been recognized as an emerging branch of 1,n-difunctionalization (Scheme 1B, c).In this context, thiosulfonates participate in the C-S formation transformations in a highly atom-economic and eco-friendly manner[22,23].With the contribution by the research groups of Huanget al.[24,25], Wanget al.[26], Zhuet al.[27,28], Zhouet al.[29],and many others [30–45], the 1,n-thiosulfonylation toolkit has been rapidly enriched during the past decades.The present review mainly highlights the recent advances in the direct construction of two distinct C-S bonds in a single operation by using thiosulfonates as dual functional reagents, namely, sulfonylating and sulfenylating reagents.Contingent on the different 1,n-thiosulfonylation modes, the focal procedures along with their scopes and suggested mechanisms are discussed in detail (Scheme 2).
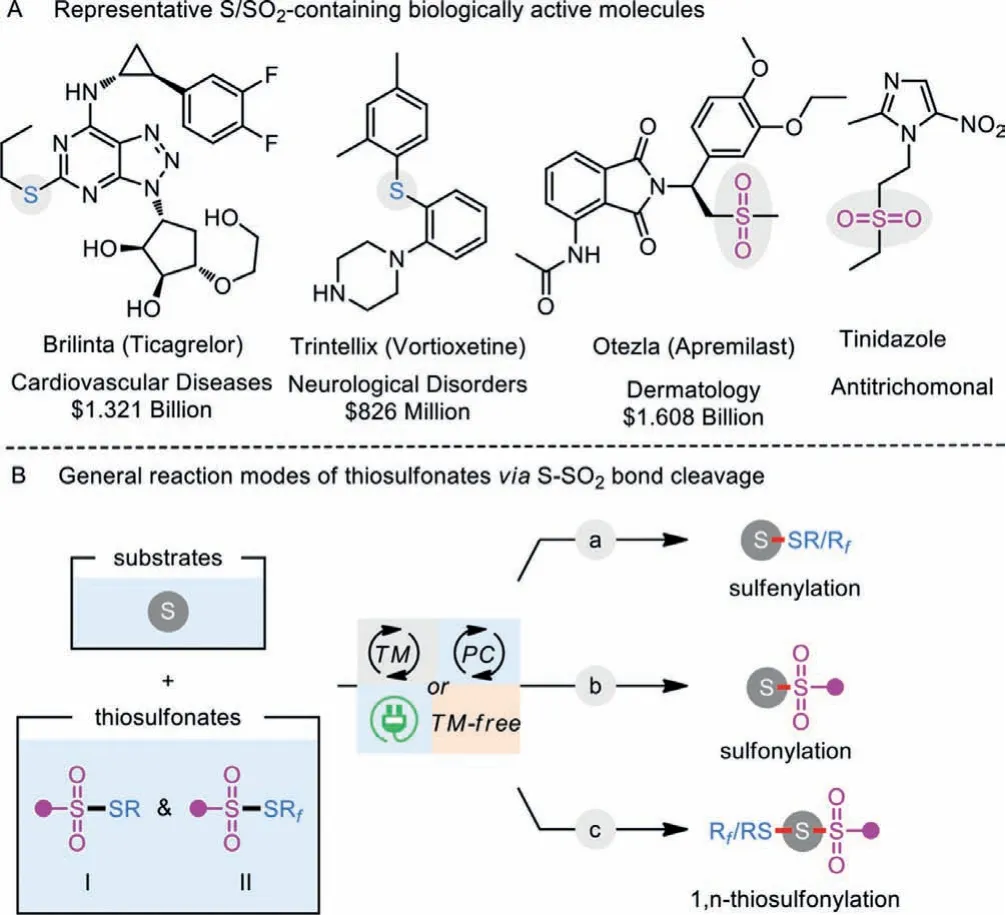
Scheme 1 .Representative S/SO2-containing drugs and general reaction modes of thiosulfonates as functional reagents.
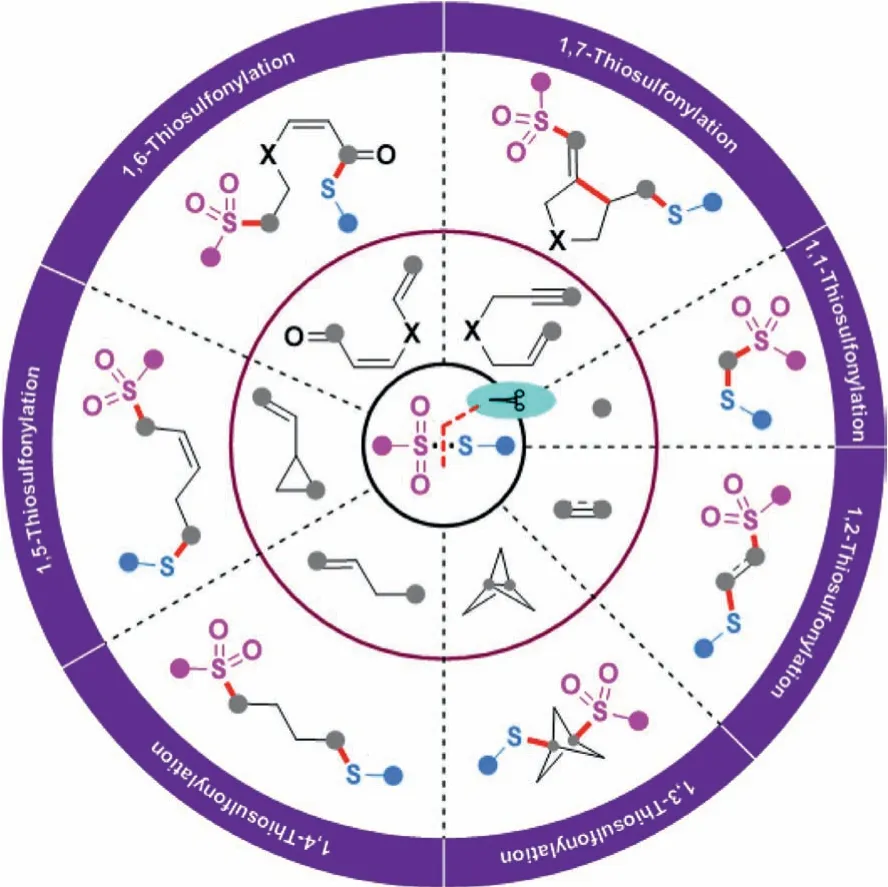
Scheme 2 .1,n-Thiosulfonylation by using thiosulfonates as dual functional reagents.
2.1,1-Thiosulfonylation
Although impressive synthetic efforts have been made in the fields of oxidative difunctionalization of olefins, the 1,1,2-trifunctionalization remains a great challenging task.In 2018, Xu’s group discovered a cascade reaction of aerobic oxidative alkene functionalization and 1,1-thiosulfonylation toβ-thio ketones from styrenes and benzenesulfonothioate under copper catalysis(Scheme 3A) [24].The trifunctionalization reaction synergistically introduced one carbonyl group and two distinct C-S bonds in a one-pot operation.The importance of oxygen for the transformation was indicated by demonstrating that the use of other oxidants such as H2O2and 1,4-benzoquinone under the same conditions provided diminished yields.Apart from terminal alkenes,indene successfully afforded the desired ketone 3.2 in 43% yield(Scheme 3B).In addition, this protocol was compatible with a variety ofS-alkyl andS-aryl benzenesulfonothioates.
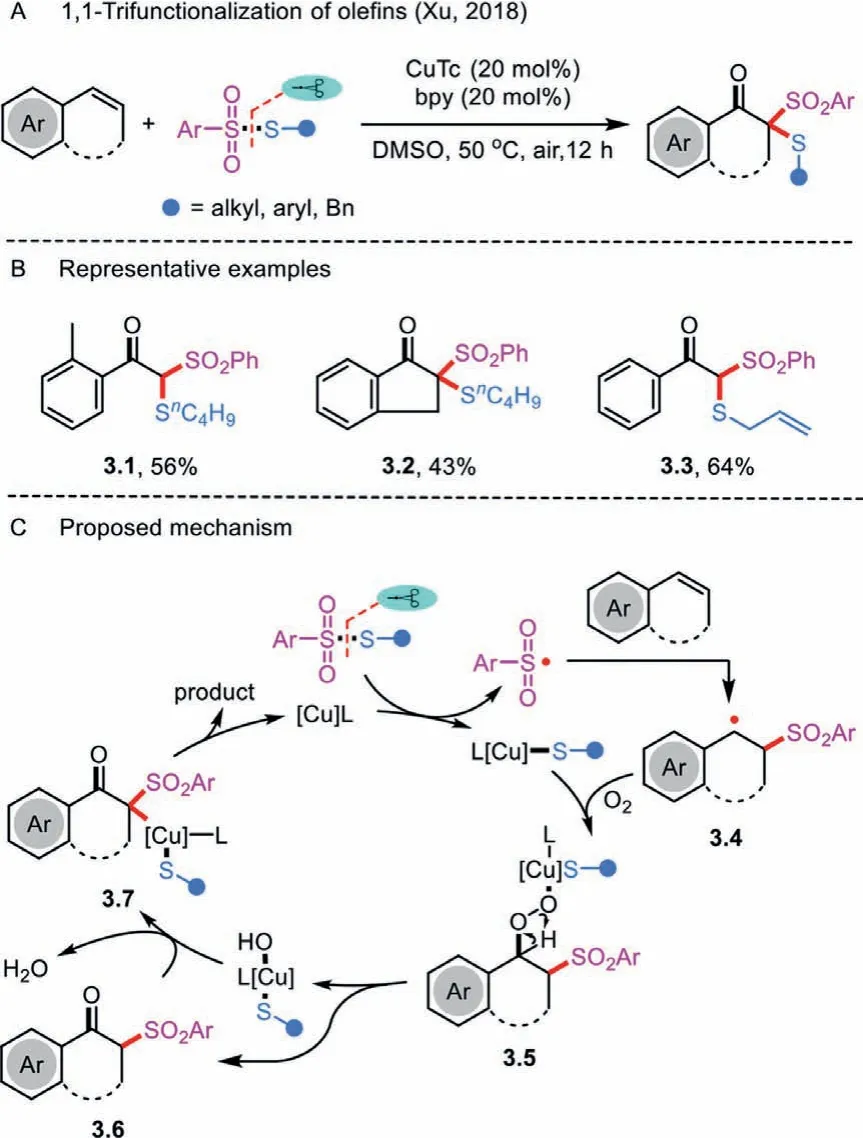
Scheme 3 .1,1-Thiosulfonylation of olefins.
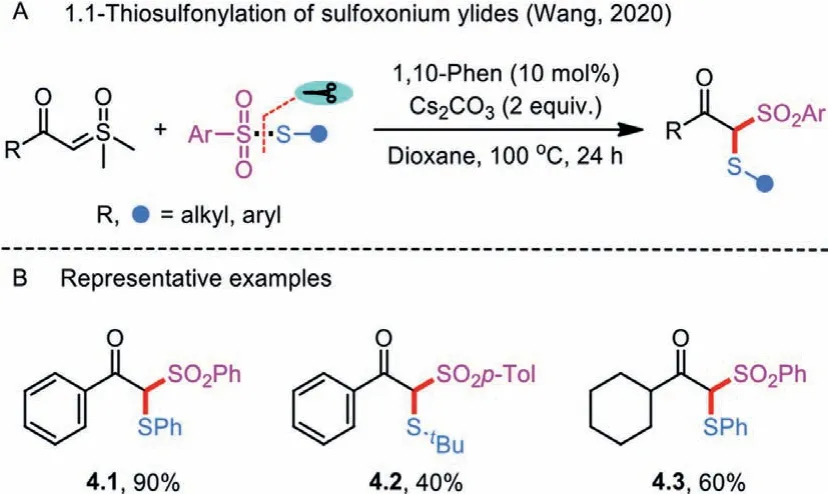
Scheme 4 .1.1-Thiosulfonylation of sulfoxonium ylides.
A copper-catalyzed mechanism is proposed in Scheme 3C.The reaction begins with the fragmentation of benzenesulfonothioate to produce an organocopper and a reactive sulfonyl radical under the action of CuTc/bpy.The latter is converted into benzyl radical 3.4 after the electrophilic radical addition to the C–C double bond of an alkene.Subsequent activation of molecular oxygen in the presence of L-Cu-S species takes place to afford the peroxy intermediate 3.5, which easily decompose to give a transientβ-keto sulfone 3.6.The interaction of the resulting 3.6 with the expelled complex L-Cu-OH generates the intermediate 3.7, which further undergoes reductive elimination to produce the desired products and regeneration of the copper catalyst.
Recently, Wang and co-workers developed a transition-metalfree insertion reaction of sulfoxonium ylides and thiosulfonates for the chemoselective synthesis ofβ-keto thiosulfones derivatives (Scheme 4A) [26].2-(Dimethyl(oxo)-λ6-sulfanylidene)-1-phenylethan-1-oneand1-cyclohexyl-2-(dimethyl(oxo)-λ6-sulfanylidene)ethan-1-one reacted well withS-aryl andS-alkyl sulfonothioates in the combination of 1,10-phenanthroline and Cs2CO3to give the corresponding products 4.1–4.3 in 40%–90%yields (Scheme 4B).
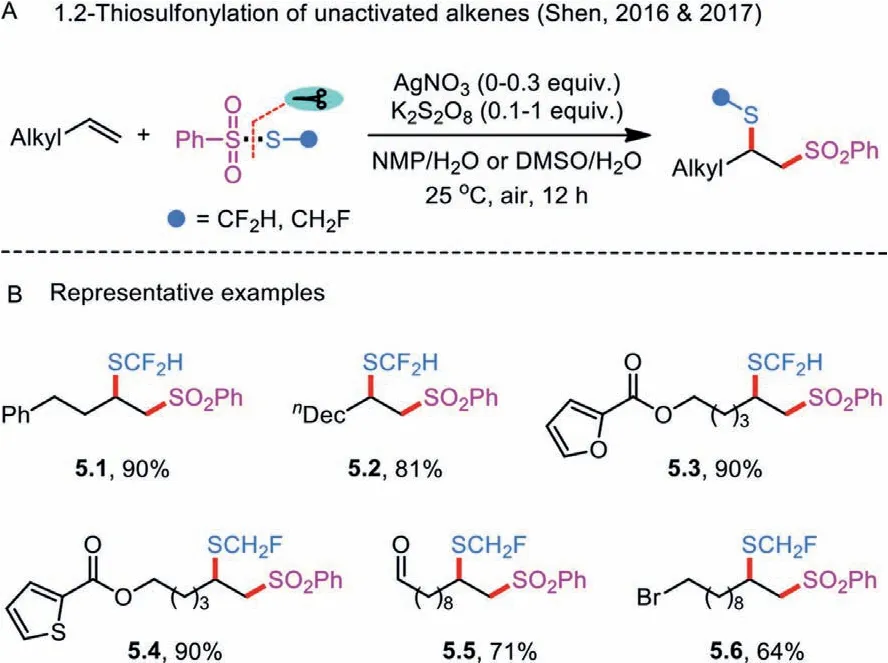
Scheme 5 .1.2-Thiosulfonylation of unactivated alkenes via silver or silver-free catalysis.
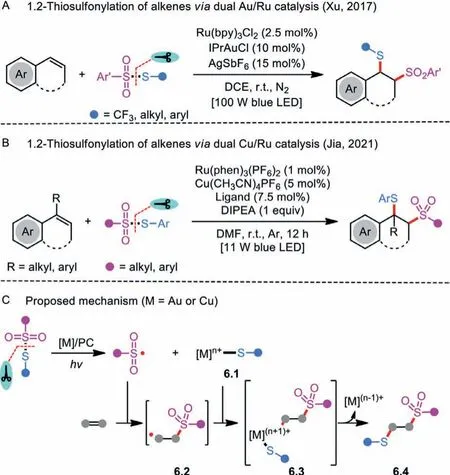
Scheme 6 .1.2-Thiosulfonylation of alkenes via metal/photoredox synergetic catalysis.
3.1,2-Thiosulfonylation
Another application of thiosulfonates is the direct regioselective 1,2-functionalization of vinyl C–C bonds in alkenes by introducing two functional handles—sulfonyl and sulfenyl moieties—at two different positions.The first 1,2-thiosulfonylation of alkenes catalyzed by silver nitrate employing stoichiometric potassium persulfate as oxidant was disclosed by Shen’s research group in 2016(Scheme 5A) [27,28].These seminal contributions allowed for the participation of monofluoromethyl and difluoromethyl benzenethiosulfonates (PhSO2SCH2F and PhSO2SCHF2) as monofluoromethylating and difluoromethylating coupling partners(Scheme 5B).The desired fluoromethylthiolated products possessing functional groups, such as carbalkoxy, formyl, and halogens,could be obtained in a highly selective manner.
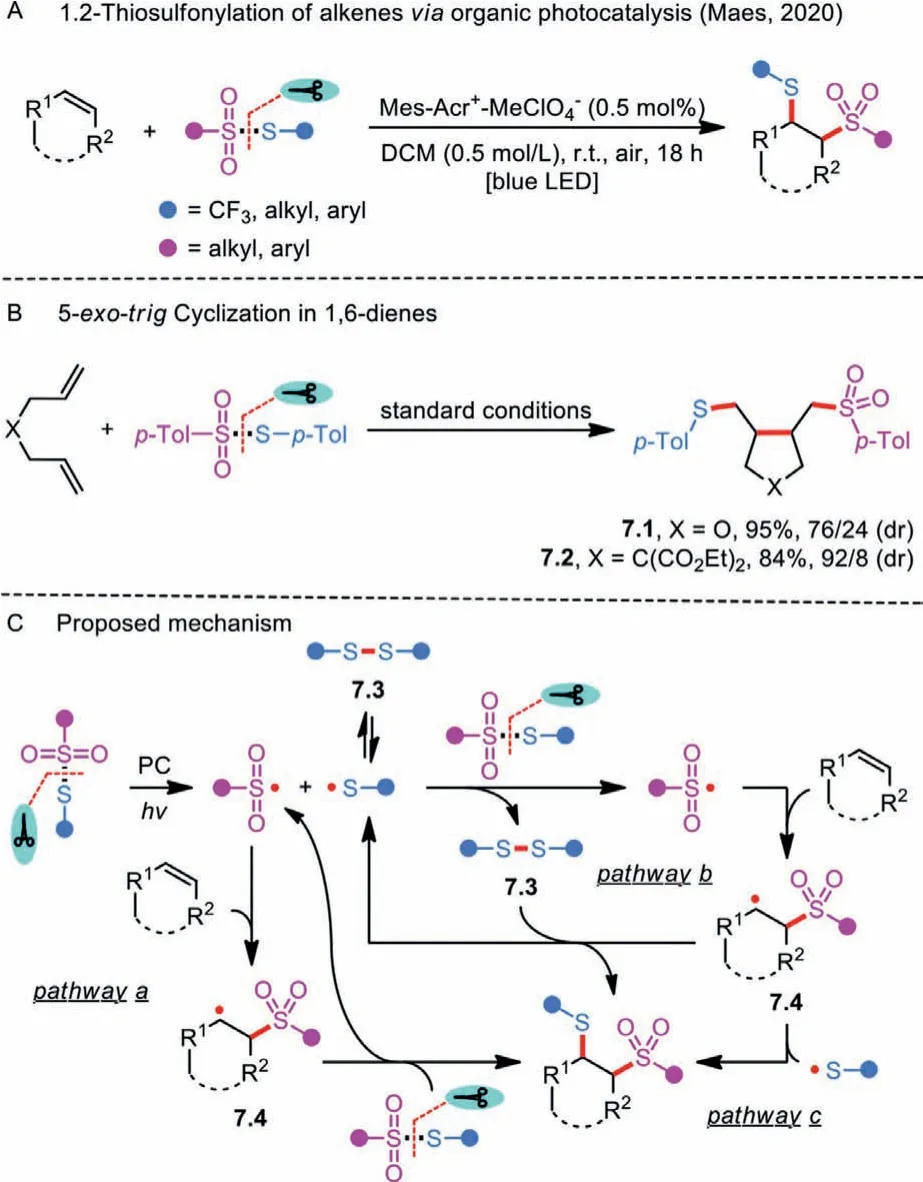
Scheme 7 .Organic photocatalyzed 1,2-thiosulfonylation of unactivated alkenes.
After that, a breakthrough in the intermolecular atom-transfer radical thiosulfonylation of alkenes was reported by Liet al.(Scheme 6A) [25].In their strategy, a dual Au (IPrAuCl) and Ru(Ru(bpy)3Cl2·6H2O) visible-light photoredox catalytic system in the presence of AgSbF6was crucial for the realization of controlled radical 1.2-thiosulfonylation of alkenes.Systematical examination of various thiosulfonate derivatives and alkene substrates found that generation of two C−S bonds with the assembly of a trifluoromethylthio group (SCF3)/thio group and a sulfonyl group into alkenes was succeeded with excellent regioselectivity and diastereoselectivity.The detailed experiment suggested that IPrAuSR was likely a central intermediate during the reaction course.
Based on the achievement, with copper salt [Cu(CH3CN)4PF6]and ruthenium-based photocatalyst [Ru(phen)3(PF6)2], a similar copper and photoredox catalysis protocol for transformingαand/orβ-substituted styrenes to valuable S-containing compounds was accomplished (Scheme 6B) [29].Jiaet al.particularly focused on the construction of benzylic quaternary carbon centers by employing the challengingα-substituted alkenes.Moreover, the practical tactic was illustrated by modifying bioactive molecules in the late stage.The mechanistic studies revealed that sulfonyl radical was involved in the reaction mixture.
Both Xu and Jia’s dual catalysis processes likely go through a photoredox-catalysis pathway rather than a classic radical chain process (Scheme 6C) [3,21].Initially, the thiosulfonate undergoes a single-electron transfer reaction with the assistance of metal/photocatalyst, leading to a sulfonyl radical and an organometal complex 6.1.Subsequently, intramolecular radical addition of the obtained reactive sulfonyl radical to the alkene takes place to deliver an alkyl/benzyl radical 6.2, followed by recombination with 6.1 to yield the species 6.3 through a single-electron oxidative event.The resulting high-valent complex 6.3 easily undergoes reductive elimination to produce the final product 6.4, and regenerates the metal catalyst.
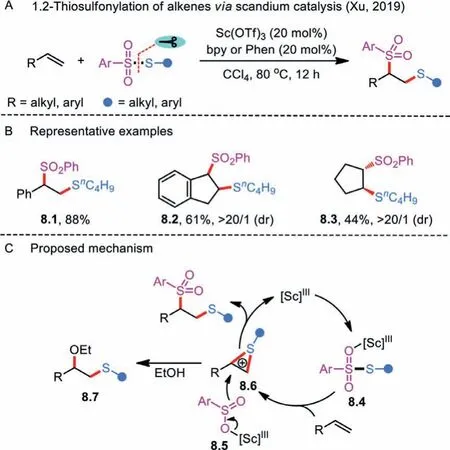
Scheme 8 .Scandium-catalyzed 1,2-thiosulfonylation of alkenes.
Meanwhile, Maes and co-workers aimed to improve the functional group compatibility by utilizing 9-mesityl-10-methylacridinium perchlorate (Mes-Acr+-MeClO4−) as a metal-free,oxidant-free, cheap, and environmentally benign photocatalyst under the irradiation of visible light (Scheme 7A) [30].Owing to the mild reaction conditions, a vast array of unactivated alkenes could be converted into 1,2-thiosulfonated products in good yields.Unfortunately, styrenes were not tolerated because of the relatively low oxidation potentials.Interestingly, when 1,6-dienes were subjected to the reactions withS-(p-tolyl)4-methylbenzenesulfonothioate, 5-exo-trigcyclized products 7.1 and 7.2 were preferentially obtained in 95% and 84% yields, respectively(Scheme 7B).
In the reaction mechanistic studies, the researchers found that a sulfenyl and a sulfonyl radical formationviathe homolytic cleavage of S-SO2bond was induced by the energy transfer from the excited Mes-Acr+-MeClO4−, rather than by a single electron transfer(Scheme 7C).Addition of the resulting sulfonyl radical to an unreactive alkene affords a new alkyl radical 7.4, which is subsequently involved in a radical chain process through the reaction with another molecule of thiosulfonate, delivering the corresponding product and a sulfenyl radical (Pathway a).Alternatively, disulfide 7.5 might participate in the production of the final product through the reaction with intermediate 7.4 (Pathway b).Additionally, there is a third possible pathway going through a radical-radical coupling of a sulfenyl radical and an alkyl radical 7.4 (Pathway c).
Alteration of reaction pathways depends heavily on the fine tuning of the catalytic system and controlling of reactive species.In this context, Xu and co-workers successfully reversed the regioselectivity of alkene 1,2-thiosulfonylation with the assistance of a Lewis acid catalystviaan ionic reaction approach (Schemes 8A and B) [31].The Sc(OTf)3-catalyzed electrophilic difunctionalization features broad substrate scope, simple procedures, and 100%atom economy.The proposed mechanism consisting of two predominant steps is outlined in Scheme 8C.In Step 1, Lewis acid Sc(OTf)3coordinates with arylsulfonothioate to ensure that the activated complex 8.4 could react with an alkene, furnishing the thiiranium ion 8.6.In Step 2, a selective attack of the arylsulfonyl anion to the congested position of intermediate 8.6 occurs to give the difunctionalized product and regenerate the Sc(III) catalyst.It is also worth mentioning that other nucleophiles may compete with ArSO2−to attack thiiranium ion.The authors observed that EtOHadducts 8.7 were formed when 10 equivalents of ethanol were added to the reaction system.Compared with established radical procedures for 1,2-thiosulfonylation of alkenes, this example nicely underscores the complementarity for accessing 2-phenylsulfonyl sulfane derivatives.
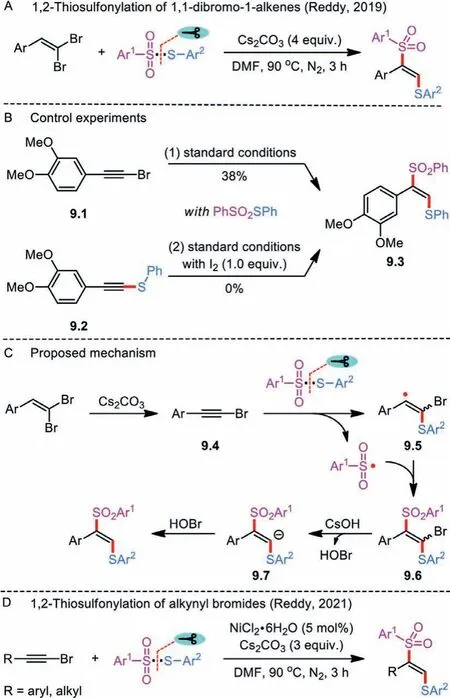
Scheme 9 .Cs2CO3-Mediated 1,2-thiosulfonylation of 1,1-dibromo-1-alkenes.
Besides simple alkenes, radical thiosulfonylation of 1,1-dibromo-1-alkenes with thiosulfonates in the presence of cesium carbonate was achieved by Reddy and co-workers (Scheme 9A) [32].The transition-metal-free reaction enabled the efficient synthesis ofαaryl-β-thioarylvinyl sulfones in moderate to excellent yields.In a control experiment, 1-bromoalkyne 9.1 was smoothly converted to the coupled product 9.3 in 38% yield under the standard conditions (Scheme 9B).In contrast, the intermediacy of thioalkyne 9.2 could be ruled out in the present system because it was susceptible to react withS-phenyl benzenesulfonothioate or with sodium benzenesulfinate in the presence of iodine.
Mechanistically, reaction initiation occurs through the basemediated dehalogenation of 1,1-dibromo-1-alkene to release alkynyl bromide 9.4 (Scheme 9C).Meanwhile, thermal homolytic scission of S-SO2bond in thiosulfonate results in the generation of a sulfonyl and a sulfenyl radical species, both of which add across the alkynyl bromide to yield an unstable compound 9.6.The final product is formed after a sequence of debromination and protonation of bromovinyl benzene 9.6.Base on this finding, the same group later demonstrated a nickel-catalyzed difunctionalization of alkynyl bromides with thiosulfonates as an alternative means to access synthetically usefulα-aryl-β-thioarylvinyl sulfone derivatives (Scheme 9D) [33].
Although there is a superficial similarity between alkynes and alkenes, the atom-transfer radical addition of alkynes has been less developed.That is because the incipient alkenyl radicals formed from radical addition to alkynes are much reactive than the alkyl radicals which generated from alkenes.Another effi-cient 1,2-thiosulfonylation method relying on an intermolecular atom-transfer addition of simple alkynesviathe combination of visible-light photoredox catalysis and gold catalysis was reported by Xu’s group (Scheme 10A) [34,35].Impressively, the radical relay chemistry permitted rapid access to diverse trifluoromethylthioand difluoromethylthio-derived vinylsulfones (Scheme 10B).This methodology was also applied to the modification of representative biologically relevant molecules (e.g., product 10.3).
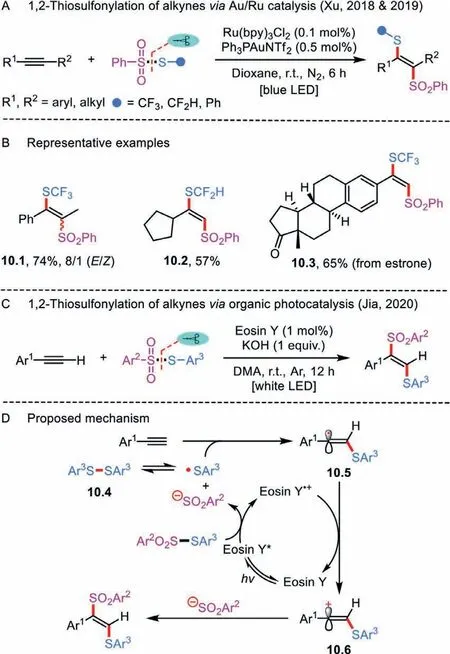
Scheme 10 .1,2-Thiosulfonylation of simple alkynes.
Soon afterward, an organic dye (Eosin Y) was chosen as an ideal photocatalyst for achieving the 1,2-thiosulfonylation of aryl alkynes, which resulted in a fully transition-metal-free process toα-thioaryl-β-arylvinyl sulfones (Scheme 10C) [36].A series of terminal alkynes containing functional groups were compatible with the mild reaction conditions.From a mechanistic point of view, the alkyne difunctionalization starts with the oxidative quenching of the excited-state Eosin Y∗by a thiosulfonate to give the sulfenyl radical, Ar2SO2anion, and Eosin Y•+species (Scheme 10D).Upon the addition of sulfenyl radical to the terminal position of alkyne,the reactiveα-alkenyl carbon radical 10.5 is formed.Subsequently,the single-electron oxidation of intermediate 10.5 by Eosin Y•+followed by an electrophilic reaction with Ar2SO2anion affords the final product.In the final stage, the Eosin Y•+is reduced to Eosin Y to close the photocatalytic cycle.
In 2020, Reddy’s group disclosed an unprecedented and unique phenylboronic acid-catalyzed dimerization ofS-benzyl thiosulfonates (Scheme 11A) [37].A wide range of benzyl disulfanylsulfone derivatives were synthesized by a sequence of tandem S–S and C–S bonds formation events under metal-free conditions(Scheme 11B).Moreover, the incorporated sulfone moiety had the potential to improve the parent molecules’biological properties.In the speculated plausible mechanism, the reaction between phenylboronic acid andS-benzyl thiosulfonate would furnish a B–S complex 11.4 (Scheme 11C).Upon heterolytic cleavage of S-SO2bond and benzylic proton abstraction in the presence of K2CO3,the excepted sulfinic acid 11.5 and a boron-sulfonium zwitterion 11.6 were obtained from complex 11.4.The latter may further interact with sulfinic acid to yield key intermediate 11.7, which attacks the sulfur moiety of another thiosulfonate to form the S-S bond coupled product.Meanwhile, phenylboronic acid could be regenerated for the next catalytic cycle.
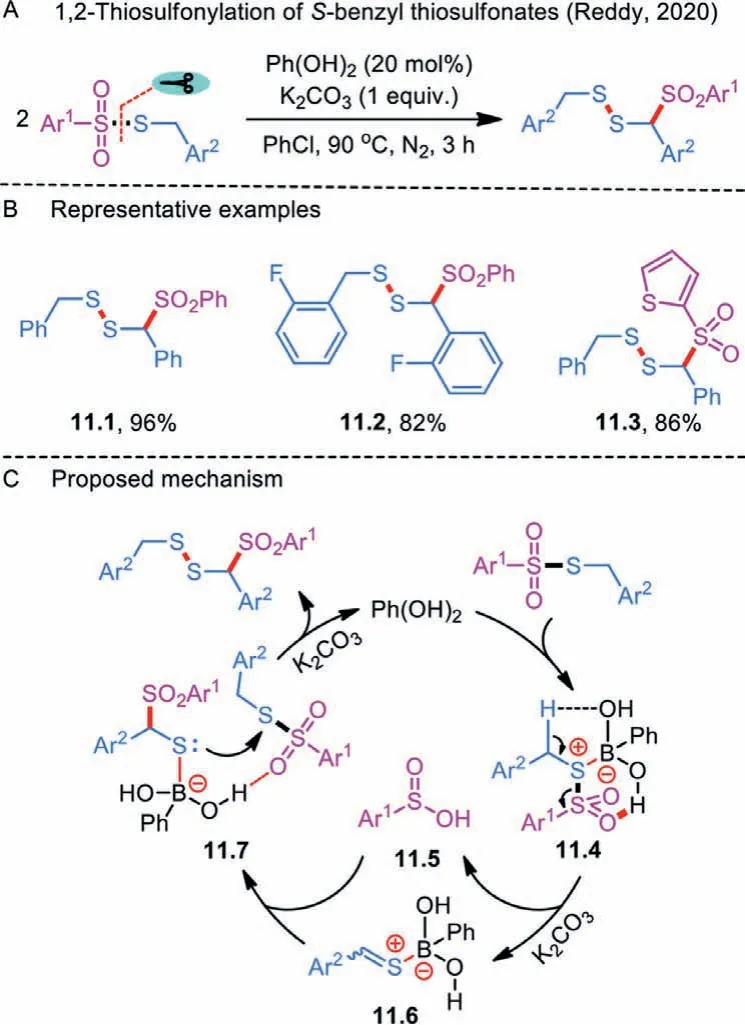
Scheme 11 .1,2-Thiosulfonylation of S-benzyl thiosulfonates.
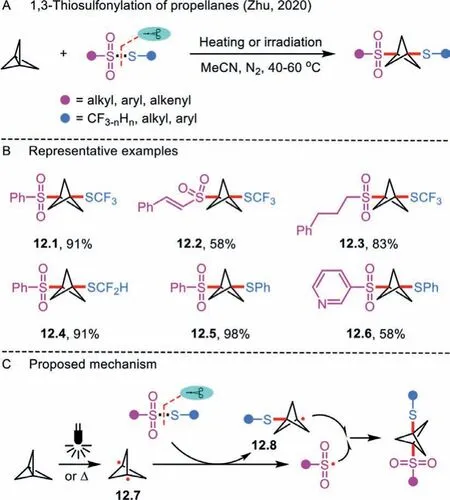
Scheme 12 .1,3-Thiosulfonylation of propellanes.
4.1,3-Thiosulfonylation
Bicyclo [1.1.1]pentane has emerged as a high-value bioisostere to enhance the chemical and physical properties of privileged molecules [38].Recognizing the need in this research field,Zhu’s group explored a 1,3-thiosulfonylation of propellanes to assemblefluoroalkylthio-functionalizedbicyclo[1.1.1]pentanes(Scheme 12A) [39,40].The thiol part in thiosulfonates could be expanded to SCF3, SCF2H, SCFH2, SAr, and S-alkyl along with SeCF3, SeC4F9, and SeC8F17analogues (Scheme 12B).Notably, the 1,3-difunctionalization [41] was also amenable to the styrylsulfonyl-substituted 12.2 or alkylsulfonyl-substituted thiosulfonates 12.3.In the mechanistic section, the author supported a radical process (Scheme 12C).The reaction goes through a diradical intermediate 12.7 upon the homolysis of the fragile central bond of propellane.Subsequently, the reaction of the obtained intermediate 12.7 with thiosulfonate generates radical 12.8 and sulfonyl radical, which subsequently couples with each other to furnish the desired product.
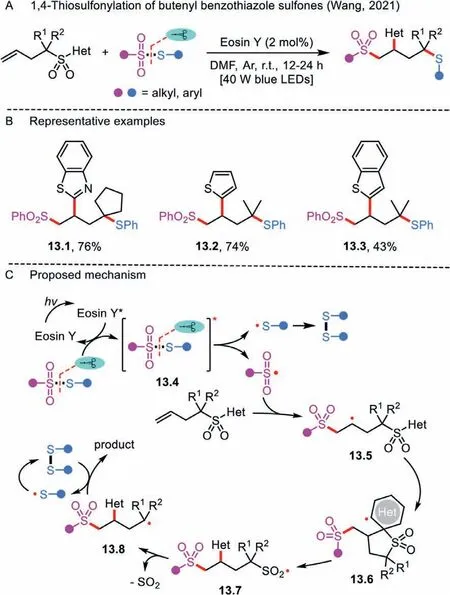
Scheme 13 .1,4-Thiosulfonylation of butenyl benzothiazole sulfones.
5.1,4-Thiosulfonylation
Radical-mediated distal aryl migration strategies have been rarely established.Very recently, a visible-light-induced sulfonylation/aryl migration/desulfonylation and C–S bond formation reaction was demonstrated by Wang’s group, thus merging radical sulfonyl-Smiles rearrangement [3] and 1,4-thiosulfonylation for the first time (Scheme 13A) [42].Importantly, this protocol was compatible with a class of butenyl benzothiazole sulfones and various thiosulfonates (Scheme 13B).However,S-ethylethanesulfonothioate was not a suitable substrate for the transformation.
The reaction is triggered by the energy transfer of thiosulfonate with the photoexcited Eosin Y∗, giving rise to a sulfenyl radical and a sulfonyl radical (Scheme 13C).The disulfide is easily formed by the homocoupling of two sulfenyl radicals.Furthermore, the addition of sulfonyl radical to the C–C double bond of the butenyl benzothiazole sulfone affords a reactive alkyl radical 13.5, which in turn undergoes intermolecular heterocyclic ring migration and SO2extrusion, leading to a tertiary alkyl radical 13.8.The stability of the resulting radical 13.8 is a promising factor for the success of the trifunctionalization.Finally, the intermediate 13.8 is intercepted by the disulfide to form the corresponding product and a sulfenyl radical.
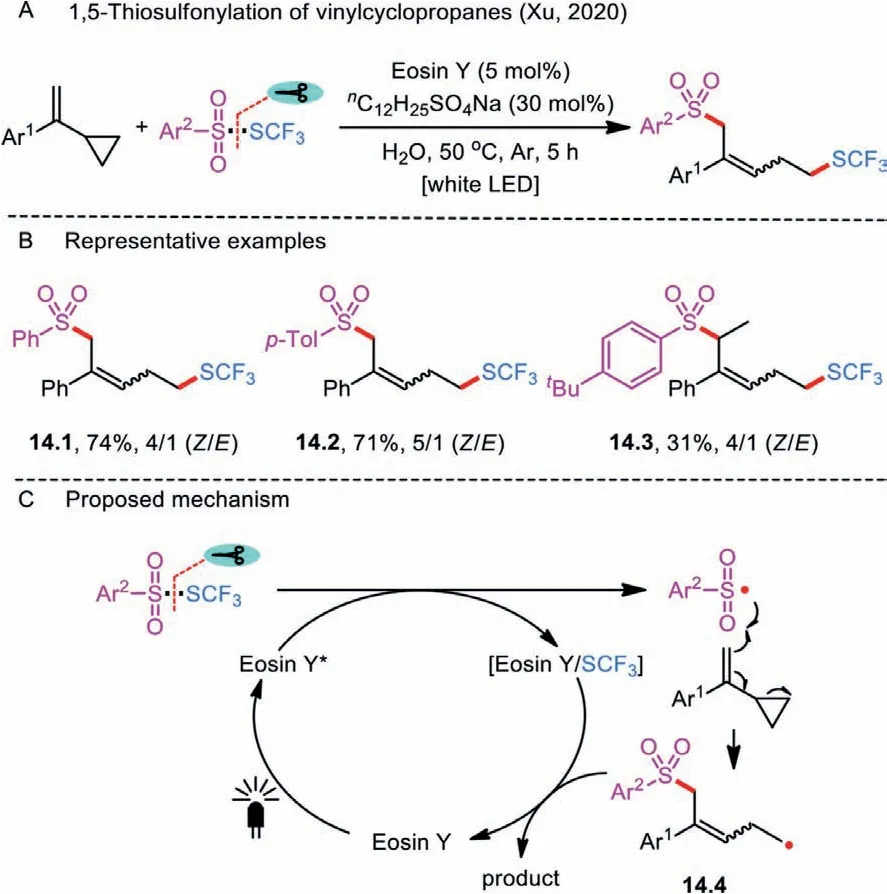
Scheme 14 .1,5-Thiosulfonylation of vinylcyclopropanes.
6.1,5-Thiosulfonylation
Ring opening of vinylcyclopropanes has been widely studied under visible-light irradiation.In 2020, an Eosin Y-mediated remote 1,5-trifluomethylthio-sulfonylation had also been accomplished through photoredox ring-opening reactions of aryl vinylcyclopropanes withS-trifluoromethyl sulfonothioate derivatives(Scheme 14A) [43].In their case, thiosulfonates served as both the SCF3and a sulfonyl source, and water was used as an environmentally-friendly solvent.It should be noted that the sodium dodecyl sulfate additive was important for increasing the solubility of organic substrates in the water phase.Both the yields and stereoselectivities of the products were acceptable under the mild reaction conditions (Scheme 14B).
The existence of radical intermediates was confirmed by the TEMPO trapping experiment.A possible mechanism for the formation of a 1,5-difunctionalized product is depicted in Scheme 14C.The catalytic cycle is initiated by the interaction of excited Eosin Y∗and Ar2SO2SCF3, affording an eosin Y/SCF3complex and an aryl sulfonyl radical.Further addition of ArSO2•to the vinyl moiety of radical acceptor followed by a ring-opening process of cyclopropane produces the unstable alkyl radical 14.4, which reacts with Eosin Y/SCF3to deliver the targeted product and releases the organocatalyst for the next catalytic cycle.
7.1,6-Thiosulfonylation
Following this strategy, Zhuet al.successfully realized a visiblelight photocatalyzed remote 1,6-thiosulfonylation of vinylaldehydes (Scheme 15A) [44].The intermolecular tandem sequence of sulfonylation and sulfenylation was triggered by the radical sulfonylation of alkenyl groups in vinylaldehydes with thiosulfonates.6- or 7-Sulfonylated thioesters with broad substrate scope were prepared in moderate to good yields by employing vinylsubstituted aryl aldehydes andα,β-unsaturated aldehydes as substrates (Scheme 15B).Of note was that hex–5-enal (product 15.3)was identified as an unsuitable variant for the transformation.Their mechanistic hypothesis showed that the formation of sulfonyl radical under the visible-light irradiation was likely the key step (Scheme 15C).In addition, the Ir-catalyzed electron transfer and homolysis of the S-SO2bond are involved in the initiation step.Addition of electrophilic sulfonyl radical to the C–C double bond of vinylaldehyde, following an intramolecular 1,5 or 1,6-H shift, leads to the generation of acyl radical 15.5.Moreover, the incipient thiyl anion could be oxidized by Ir(V) species to yield a sulfenyl radical,which attacks the resulting 15.5 to form the targeted thioester.
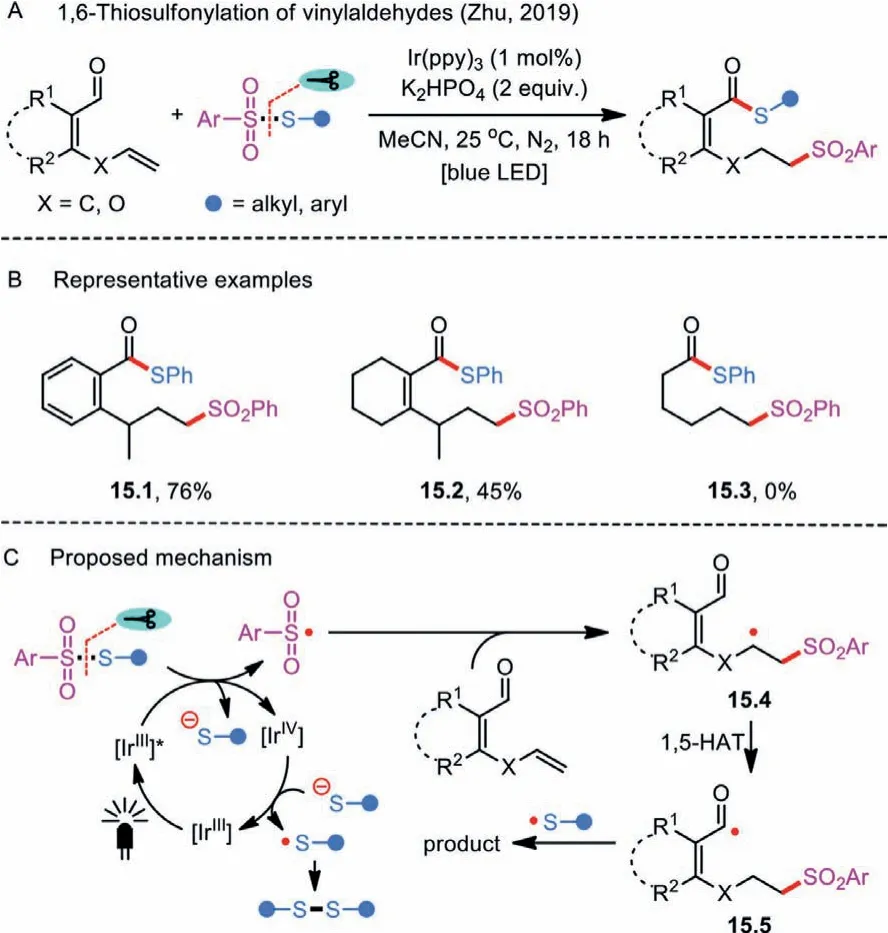
Scheme 15 .1,6-Thiosulfonylation of vinylaldehydes.
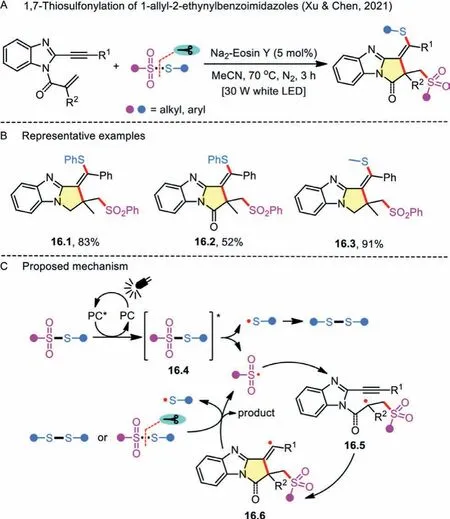
Scheme 16 .1,7-Thiosulfonylation of 1-allyl-2-ethynylbenzoimidazoles.
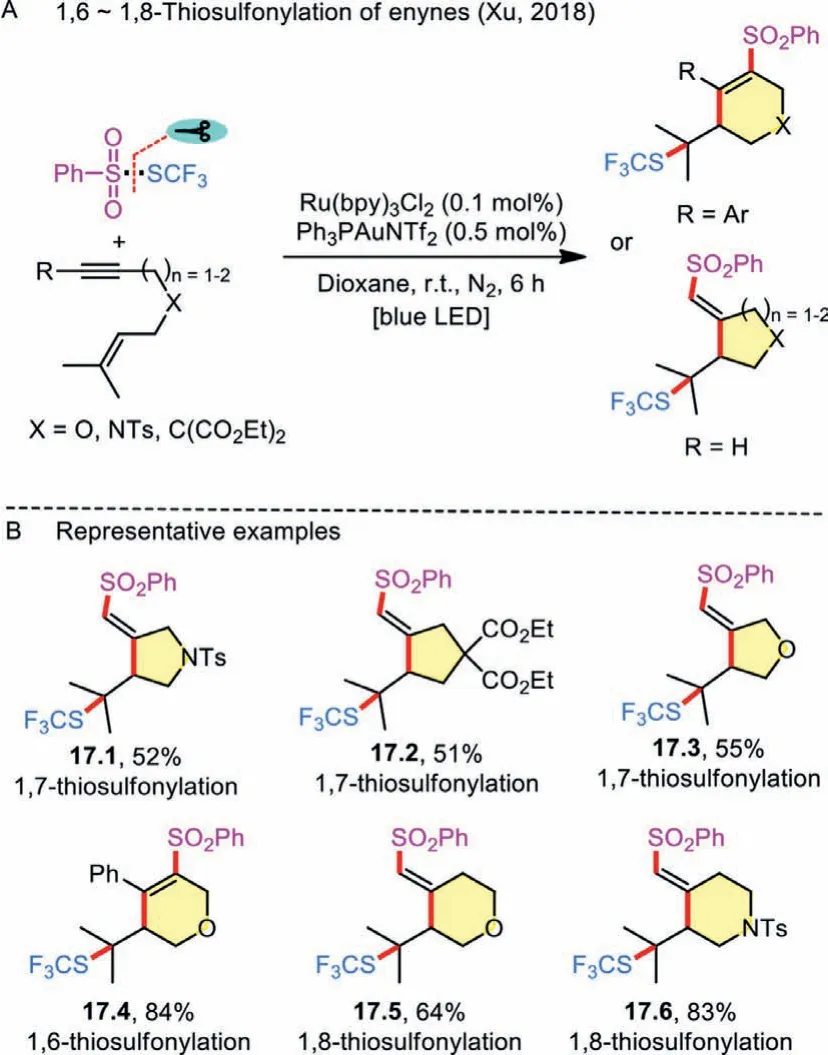
Scheme 17 .1,n-Thiosulfonylation of enynes.
8.1,7-Thiosulfonylation
On the other hand, radical domino reaction enables the facile construction of novel heterocycles.By virtue of the 1-allyl-2-ethynylbenzoimidazole as a radical acceptor, the 1,n-thiosulfonylation protocol was further applied to the transition-metal-free synthesis of thiosulfonylated pyrrolo[1,2-a]benzimidazoles (Scheme 16A) [45].The broad functional group tolerance and potential application in the manufacture of functionalized materials was another merit of the method (Scheme 16B).A similar aforementioned photocatalytic mechanism for organic dye-mediated cascade reaction was also proposed (Scheme 16C).First, Na2-Eosin Y is activated to the excited state under the irradiation of white LEDs.Second, the energy transfer between the photoexcited catalyst with thiosulfonate results in a sulfonyl radical and a sulfenyl radical through excited intermediate 16.4.Subsequent addition of the resulting sulfonyl radical to 1-allyl-2-ethynylbenzoimidazole generates radical intermediate 16.5, which further undergoes intramolecular cyclization to result in an alkenyl radical 16.6.Finally, the intermediate 16.6 is attacked by disulfide or thiosulfonate to produce the corresponding product, and the sulfonyl radical is simultaneously regenerated.
Thiosulfonylation of 1,n-enynes can also be used for constructing specific ring skeletons.In this regard, Xu and co-workers tested the gold/photocatalyst-mediated cascade cyclization of enynes to access five- or six-membered carbo- and heterocycles by 1,6-/1,7-/1,8-thiosulfonylation (Scheme 17A) [34].The ability of the strategy to enable the convenient conversion of a variety of oxygen- or nitrogen-linked enynes to thio-functionalized heterocycles is attractive, demonstrating its superiority to the traditional heteroannulation (Scheme 17B).
9.Conclusions
The recent advances of 1,n-thiosulfonylation by using thiosulfonates as dual functional reagents during the past several years have been summarized and classified on the basis of different reaction modes.A series of unsaturated substrates, including alkenes,sulfoxonium ylides, alkynes, propellanes, butenyl benzothiazole sulfones, vinylcyclopropanes, vinylaldehydes, and enynes, are functionalized by selectively installing sulfonyl and sulfenyl groups at 1- andn-positions of substrates, which inarguably provides chemists an alternative strategy for the construction of highly functionalizedS-containing compounds.In these manners, thiosulfonates are generally incorporated in the desired products in 100% atom economy in accordance with the requirements of green chemistry.Moreover, this review has highlighted the generality of substrate scope and the details of suggested mechanisms.
Although significant progress has been made in the 1,nthiosulfonylation with the employment of thiosulfonates as versatile building blocks, many problems are still unsolved in this research topic.From the viewpoint of green chemistry, the development of a new catalytic techniqueviathe replacement of metal catalysts with common metal-free promoters will be impactful, especially in the diverse synthesis of bioactive products and drugs.Furthermore, continuing efforts in their utility in multi-component and tandem reactions by using photocatalysis and electrocatalysis have the potential to rapidly build up molecular complexity.In addition, there is a high demand for achieving catalytic asymmetric versions of the present 1,n-thiosulfonylation transformations.Finally, we hope this review might inspire researchers to discover new types of 1,n-thiosulfonylation-relevant reactions in this rapidly growing area.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We gratefully acknowledge the financial support from the Natural Science Foundation of China (No.22001121), Natural Science Foundation of Jiangsu Province (No.BK20180690), and Nanjing Tech University (Start-up Grant Nos.39837118 and 39837146), and Xuzhou Medical University (Start-up Grant No.RC20552038).
 Chinese Chemical Letters2022年11期
Chinese Chemical Letters2022年11期
- Chinese Chemical Letters的其它文章
- Zeolite-based Fenton-like catalysis for pollutant removal and reclamation from wastewater
- Degradation of florfenicol in a flow-through electro-Fenton system enhanced by wood-derived block carbon (WBC) cathode
- Simultaneous determination of indole metabolites of tryptophan in rat feces by chemical labeling assisted liquid chromatography-tandem mass spectrometry
- Self-powered anti-interference photoelectrochemical immunosensor based on Au/ZIS/CIS heterojunction photocathode with zwitterionic peptide anchoring
- The role of Cs dopants for improved activation of molecular oxygen and degradation of tetracycline over carbon nitride
- Heterostructures of NiFe LDH hierarchically assembled on MoS2 nanosheets as high-efficiency electrocatalysts for overall water splitting