
合金元素对Ag/Al界面性质影响的第一性原理研究
2022-12-07 09:36冉小杰黄福祥曾利娟徐良玉
原子与分子物理学报 2022年1期
冉小杰, 周 露, 黄福祥, 曾利娟, 徐良玉
(重庆理工大学 材料科学与工程学院, 重庆400054)
1 引 言
键合丝是微电子封装业的重要结构材料之一,其作用是连接半导体芯片与基板,使芯片与外界电气连接、电信互通[1, 2]. 随着电子封装向小型化、封装多引线化、模块化发展,对键合丝提出了更高的要求. Ag具有优异的导电、导热性能和良好的力学性能,已发展成为除了Cu丝外的另一种替代Au丝的基础材料[3]. 纯Ag丝本身强度较低,在高速键合条件下易断线和硫化;此外,高纯Ag丝和Al焊盘易形成金属间化合物,会增加键合界面的电阻抗和热阻抗[4, 5],目前通过微合金化技术是改善以上问题的主要方法,通过加入少量的合金元素能有效的改善银丝的抗拉强度和拉丝性能以及键合可靠性等[1,6, 7].
在微连接领域中,Ag/Al异质界面连接是最为常见的结合方式. 据统计,由界面引起器件失效的形式占整个电子封装可靠性问题的25%[8],由此Ag/Al界面的结合是保证器件服役寿命的关键. 实验研究发现,微合金化元素对Ag/Al界面结合有显著影响[9-13]. 如Tseng[9]指出Ag基体中添加少量的Pd或Au能抑制Ag/Al界面间金属间化合物的生长,有效提高结合性能;康菲菲[10]发现Ag丝中掺杂Cu、In和Ce不仅具有较高的力学性能,而且提高了界面Ag+的电迁移电阻;Lin[11]介绍了一种含有微量Au、In、Ni的Ag-Pd合金丝经高温储存寿命试验(HTST)和温度循环试验(TCT)后Ag/Al界面仍具有良好的可靠性. 然而,由于界面微观结构的复杂性和观测手段的限制,其中关于界面的微观结合机理及微合金化元素的影响规律还有待探讨.
基于密度泛函理论的第一性原理计算方法已被证实是研究材料的物理、化学性质的有力工具,能从原子尺度为界面性质的认识提供全新的思路. 近年来,金属/金属界面的第一性原理计算为揭示界面的成键本质和原子结构做出了重要贡献. 如彭艳[14]、赵磊[15]、徐沛瑶[16]等人采用第一性原理计算方法讨论了合金元素对(Cu,Al)/Fe界面结合性质的影响,研究发现合金元素的添加对界面结合强度有不同程度的影响. 此外,关于从能量学角度判断Ag/CuO[17]和Ag/Ni[18]界面稳定性的第一性原理计算也有报道. 因此,本文从界面能量学、电子结构角度探讨合金元素(Be、Mg、Al、Ni、Ca、Sn)添加在Ag中对Ag/Al界面的影响,给出了界面能、分离功、态密度、差分电荷密度以及布居分析等计算结果,从中探索其变化规律及机制,为新型银键合丝的研究与生成提供指导作用.
2 计算方法与结构模型
2.1 计算方法
本文采用基于密度泛函理论(DFT)的CASTEP(Cambridge Sequential Total Energy Package)[19]软件包完成对Ag/Al界面性质的第一性原理计算. 计算过程中利用广义梯度近似(GGA)下的Perdew-Burke-Ernzerhof (PBE)泛函[20]来处理体系中电子与电子交换关联能部分. 采用超软赝势(Ultrasoft)描述离子实与价电子之间的相互作用,并通过自洽迭代法(SCF)进行收敛性计算[21],收敛精度设置为1.0×10-6eV/atom. 为了满足计算的精度并提高计算速度,测试了体系总能量与平面波截段能和K点网格划分的收敛性. 经确定在Ag和Al的体相和表面性质计算过程中,平面波截断能分别采用为350 eV 和300 eV ,布里渊区Monkhorst-Pack[22]取样积分为7×7×7和5×5×5. Ag/Al界面性质计算过程中平面波截断能选择为400 eV,布里渊区k点划分为5×5×2. 迭代自洽收敛条件分别为:总能量小于1.0×10-5eV/atom,原子平均受力低于0.3 eV/nm,公差偏移小于0.01 nm,最大应力偏差为0.05 GPa.
2.2 模型建立
课题组前期对Ag/Al界面稳定的几何结构进行了详细探讨,结果表明以密排面表面构建的Ag(111)/Al(111)界面最为稳定[23]. 在此基础上,本文采用具有真空层的超晶胞建模方法,构建Ag(111)/Al(111)位向关系的界面模型. 计算过程中由于界面模型中包含的原子层数越多,计算时间将会增加,而层数较少,则计算结果的误差会偏大. 因此,分别建立了Ag(111)和Al(111)面的表面模型,考察其表面能随原子层数的收敛趋势. 随后在确定原子层数后,将两者堆垛成具有合适界面间距的界面模型,如图1(c)所示,模型中包含20个Ag原子和20个Al原子. 本文探讨了合金原子在不同Ag层中取代位置的置换能,确定合金原子在界面位置的占位倾向. 同时,计算了合金元素掺杂前后体系的界面能、分离功、界面电子结构,分析了Ag中添加的合金元素对Ag/Al界面性质的影响.

图1 (a)Ag(111)表面优化模型;(b)Al(111)表面优化模型;(c)Ag(111)/Al(111)界面模型
3 结果与讨论
3.1 体相及表面性质
为了获得精确的计算结果,首先对Ag和Al晶胞的结构进行优化,基于优化结果,再构建界面结构模型进行计算. 计算得到的平衡晶格常数和体模量列于表1,从表中发现:计算的Ag平衡晶格常数、体模量与许灿辉[24]提到的实验值和理论计算值基本吻合;同时,计算得到Al平衡晶格常数、体模量也与Siegel[25]获得的实验值和理论计算值基本一致. 说明本文采用的计算方法和参数能够保证足够的精度和准确性.

表1 Ag和Al的平衡晶格常数及体模量
界面构建过程中为保证Ag/Al界面两侧原子具有体相特征,而又不至于过厚而增加计算量,因此通过考察表面能(σ)随原子层数的收敛性来确定界面两侧的Ag和Al表面的最小原子层数. 表面能(σ)作为判断表面稳定性最重要的参数,可由以下公式计算[26]:
(1)
式中:Eslab和Ebulk分别为表面模型和块体单胞的总能量;N为表面模型中所含单胞数;A为表面总面积. 图2为计算得到的表面能与原子层数收敛性曲线. 从图中可以看出,当原子层数达到5层时,Ag(111)和Al(111)面表面能趋于收敛. 因此,后续界面的堆垛模型中Ag原子层和Al原子层均取为5层. 计算得到Ag(111)面的表面能为 0.786 J/m2,Al(111)面的表面能为0.840 J/m2,这与许灿辉[24]计算得到的Ag(111)面的表面能为0.74 J/m2和Rodriguez[27]计算得到的Al(111)面的表面能为0.89 J/m2基本吻合,说明本文的计算结果可信.
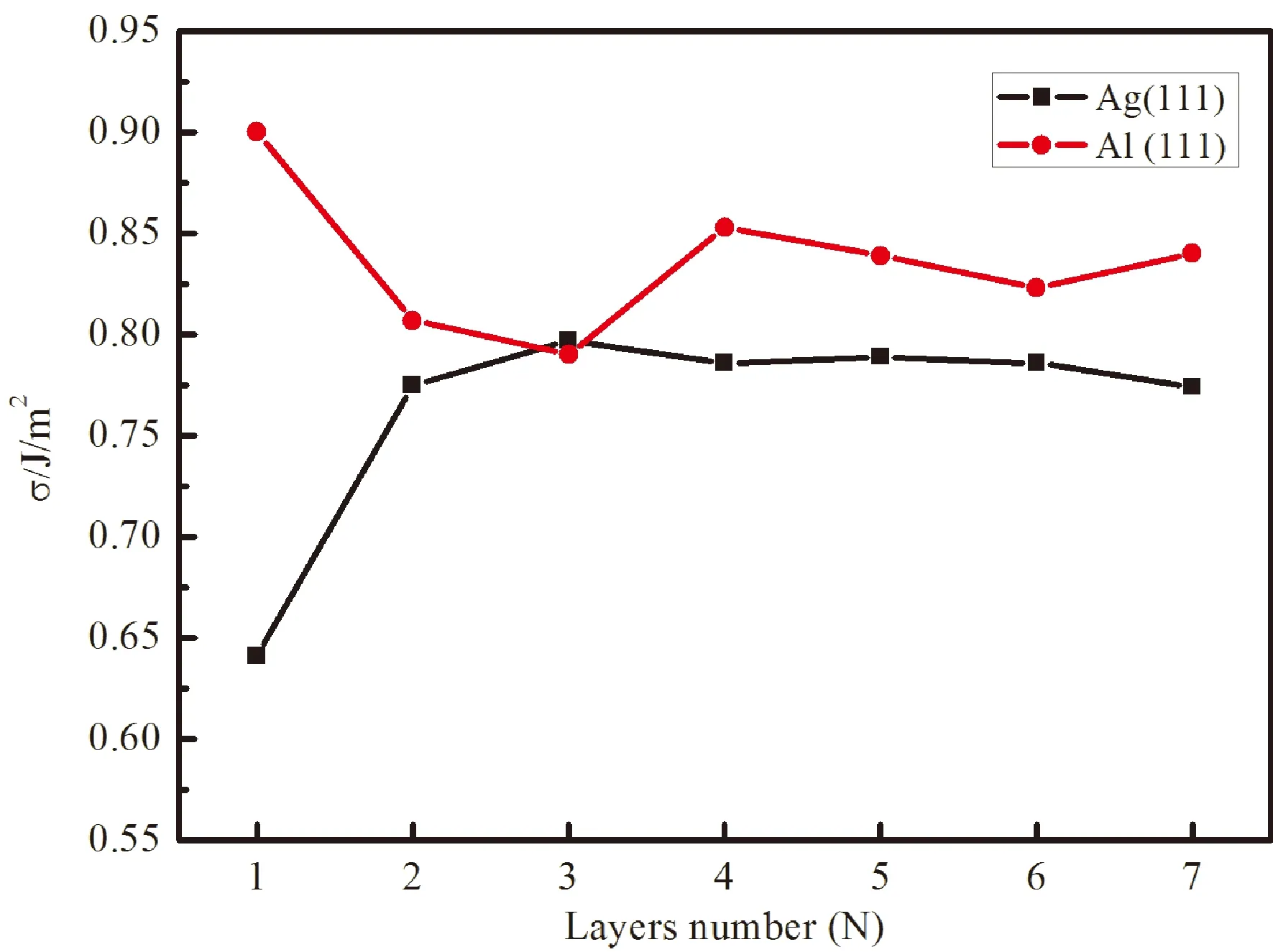
图2 表面能随原子层数的变化
3.2 合金元素在Ag侧的占位
为了研究Ag合金中添加的合金元素在Ag / Al界面上的优先占位,计算了每种合金原子在不同取代位置的置换能,其中置换能最低的位置即为合金元素最易取代的位置. 为便于说明,对模型中Ag原子的取代位置进行了标号,蓝色和粉色球分别代表Ag、Al原子,如图3所示. 置换能可以通过以下公式计算[16]:

图3 合金原子在Ag / Al界面中的不同置换位置
(2)
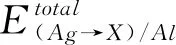
置换能的计算结果见表2,从表中可以看出Mg、Al、Ca、Sn原子在不同取代位置处的置换能均为负值,表明置换容易进行,而Be、Ni原子在不同取代位置处的置换能均为正值,表明置换相对较困难. 其中Ni原子在最靠近界面的位置1处置换能最低,Al、Sn、Ca原子在远离界面的位置5处置换能最低,而Be和Mg原子在位置3处置换能最低. 因此,后续在考虑合金元素对界面性能影响的计算中,合金原子的最佳取代位置均考虑为对应置换能最低的位置.
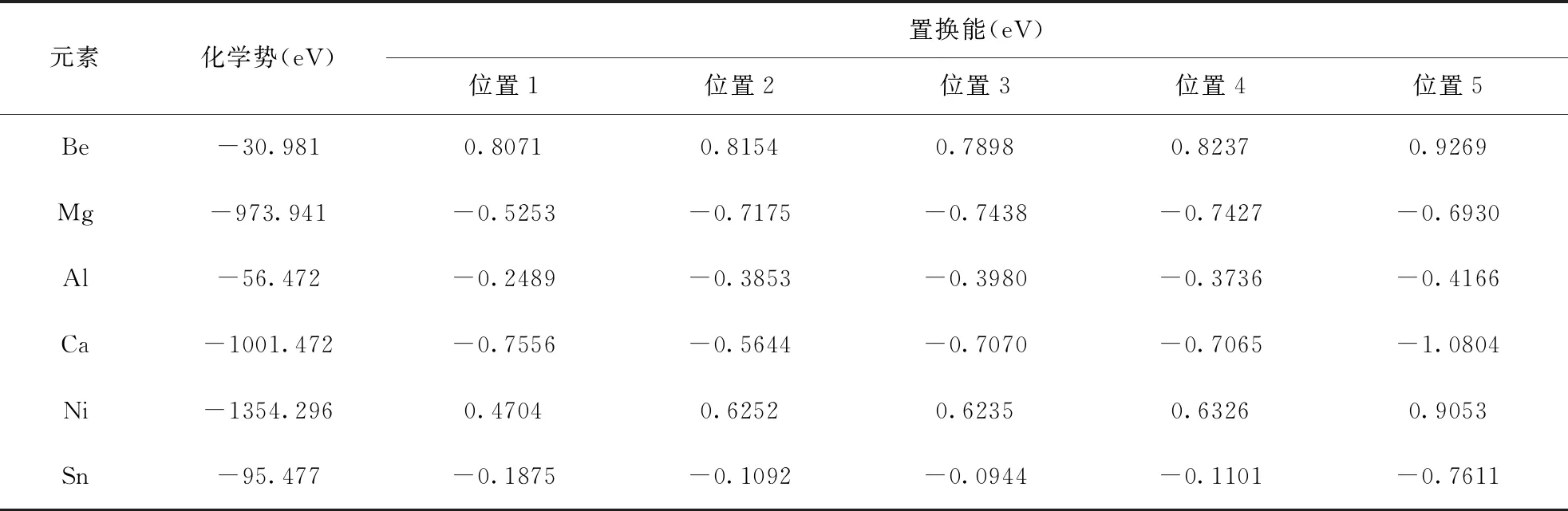
表2 合金原子在不同取代位置的置换能
3.3 界面结合稳定性
分离功(Wsep)和界面能(γint)作为两个重要的参数提供了界面强度和结合的相关信息,对于评估界面的稳定性起到了重要的作用[28].其中,分离功(Wsep)指把稳定界面分离成两个独立且距离无限远的自由表面结构所需要的能量,可以表征界面的结合强度,Wsep越大,表示将两者分离所需能量更多,因此界面结合越稳定. 可根据下式计算得到[29]:
(3)

界面能表示为两个自由表面形成完整界面后单位表面面积引起的能量差,本质来源于界面处原子畸变、化学键的改变和结构应变引起的能量差值[30]. 界面能越高,表明形成界面所需的能量越大,不利于界面形成,因此界面能越低,越易形成. 具体的计算公式如下[29]:
(4)
(5)

γint,Ag/Al=σAg+σAl-Wsep,Ag/Al
(6)
根据以上关系,计算得到的加入合金元素后Ag/Al界面的界面能和分离功见表3. 未掺杂前Ag/Al界面的分离功和界面能分别为1.378 J/m2和0.248 J/m2,表明具有良好的界面稳定性. 而随着合金原子Be、Mg、Al、Ca、Ni、Sn的加入,体系的分离功降低,分别为:1.066 J/m2、0.994 J/m2、1.042 J/m2、0.923 J/m2、1.261 J/m2、1.006 J/m2,对应的界面能增加,分别为:0.248 J/m2、0.56 J/m2、0.632 J/m2、0.584 J/m2、0.703 J/m2、0.365 J/m2、0.620 J/m2,表明合金元素的加入均不同程度的降低了界面的稳定性,其中Ni元素对Ag/Al界面稳定性影响较小,而Ca元素削弱程度较大. 由于合金原子的原子半径及电负性与Ag原子性质存在差异,因此合金原子的引入会导致Ag/Al界面的晶格常数发生畸变,从而影响界面结合,故本文考察了体系相对体积的变化对界面稳定性的影响. 体系的相对体积即合金原子加入后引起的界面体积的改变,可根据以下公式计算:

表3 不同元素掺杂后Ag/Al界面的分离功与界面能 (单位:J/m2)
ΔV=Vaf(Ag19,X/Al20)-Vbf(Ag20/Al20)
(7)
式中,Vbf(Ag20/Al20)和Vaf(Ag19,X/Al20)分别为掺杂前后Ag/Al界面的体积.
图4为掺杂前后Ag/Al界面的相对体积与分离功和界面能关系图,从图中可以看出,体系分离功基本与相对体积呈现线性负相关关系,界面能和相对体积成正比关系,即加入相对体积改变较小的元素(如Ni和Be),界面畸变程度较低,相对来说体系的稳定性和结合能力更好.

图4 界面相对体积与分离功、界面能的关系
3.4 电子结构分析
为了从微观尺度分析Be,Mg,Al等微合金化元素对Ag/Al界面结合的影响机制,计算了态密度、差分电荷密度以及布居分析,了解界面的电子结构和成键性质以及电子的空间分布.
态密度(DOS)能反应界面原子对能态的贡献,合金原子取代Ag原子前后的Ag/Al界面模型的分波态密度(PDOS)如图5所示. 从图中可以发现,合金化前后的所有Ag/Al界面中,第一层和第五层Ag之间的DOS曲线差别很大,这可能是由界面之间的相互作用引起的,这意味着Ag原子在界面和基体中的性质显著不同. 未掺杂Ag/Al界面中,如图5(a),对成键电子有贡献的能量主要集中在-6.5--2.5 eV范围内,在该范围内存在明显的重叠峰,参与杂化的电子主要有 Ag 4d、Al 3s 和 Al 3p. 合金化后,合金原子与界面间的Ag原子和Al原子相互作用,原子成键区域发生改变,如Mg、Ca、Ni元素的加入,在界面低能级区域均有成键峰,分别对应于Mg 2p、Ca 3s和Ca 3p、Ni 3d电子轨道贡献.
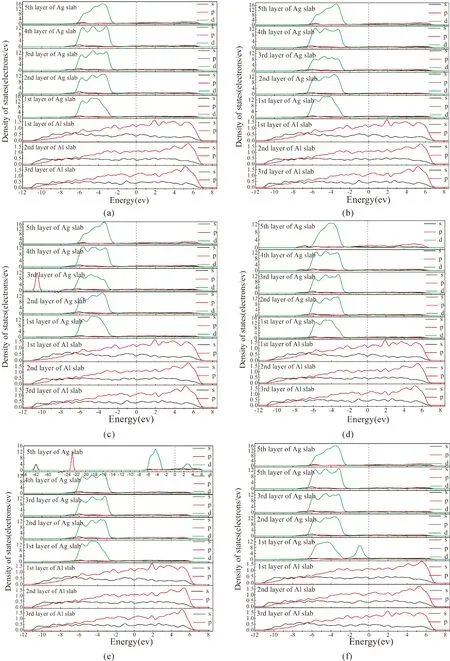
图5 Ag/Al界面的态密度图;(a)-(g)分别为:未掺杂, Be, Mg, Al, Ca, Ni, Sn掺杂体系
图6进一步给出了Be、Mg、Al、Ca、Ni元素合金化后的Ag/Al界面电荷差分密度图(均考虑为在(100)面上投影),图中蓝色区域和红色区域分别表示电荷密度减少和增加. 从图中可以看出它们均具有相似之处,即界面处第一层的Ag原子和第一层Al原子间均有电子云重叠,从而形成了较强的Ag-Al共价键. 界面中同一片层内的Ag原子和Al原子周围分布的电子表现出一定的定域性和电子共用特征,显示出Ag-Ag和Al-Al之间既有离子键又有共价键结合. 加入Be、Mg、Al、Sn、Ca后,界面处的电荷富集程度降低,原子间化学键合能力降低,形成界面所需能量更高,导致界面的稳定性被削弱. 加入Ni元素后,Ni原子周围电荷大量聚集,表明Ni原子的离子性,使相较与其他合金元素的加入的界面稳定性有所提升.

图6 Ag/Al界面差分电荷密度图
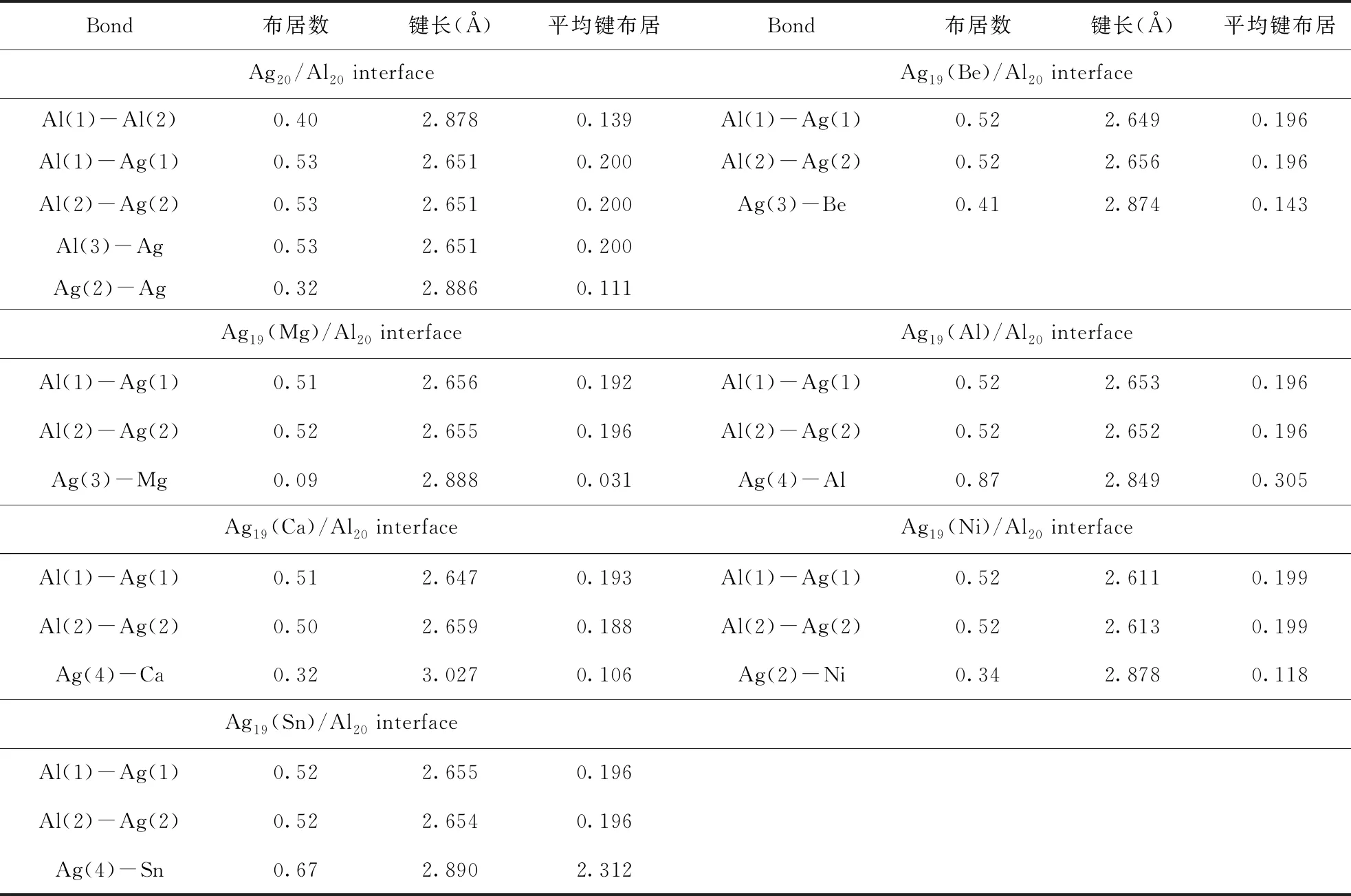
键重叠布居数的绝对值大小代表了原子间成键的强弱,该值越大,说明所成键结合能力越强;并且当键布居数为正时,成键具有共价键性,反之则为离子键. 为此,本文计算了合金元素掺杂前后Ag/Al界面附近各原子间的电荷分布及成键的重叠布居数,结果如表4所示. 其中,原子标号见图7. 从表中可以看出,未掺杂前界面上的键Al(1)-Ag(1)、Al-Al和Ag-Ag的重叠布居数分别为0.53、0.40、0.32,说明界面附近原子主要成键形式为共价键,且界面处的Ag-Al共价键强度高于两相内部结合强度,因此界面具有较好的稳定性. 而Be、Mg、Al、Ca、Sn合金元素掺杂后,键Al(1)-Ag(1)、Al(2)-Ag(2)的平均键布居值均不同程度减小,其顺序为:Be 图7 Ag/Al界面附近原子位置示意图.红球代表Ni取代位置;绿球代表Be、Mg取代位置;蓝球代表Al、Ca、Sn取代位置 表4 掺杂前后界面处各原子间的电荷分布及成键的重叠布居数 综上所述,本文采用基于平面波赝势的DFT-GGA方法研究了Ag和Al体相和它们的低指数面以及微合金化元素 (Be、Mg、Al、Ca、Ni、Sn) 对Ag/Al界面性质的影响. 本研究得出以下结论: 1)在所研究的掺杂元素中Be、Mg原子倾向于取代Ag侧靠近Ag/Al界面处的位置3,Al、Ca、Sn原子位于最远离界面的位置5,Ni原子位于最邻近界面的位置1. 2)Ag中掺杂的合金元素 均使界面的分离功减小,界面能增加,即削弱了界面稳定性. 其中,Ca原子与置换Ag原子的原子半径及电负性与Ag原子性质存在明显差异,导致界面相对体积增加最大,界面畸变程度严重,故Ca掺杂对Ag/Al界面稳定性减低最为严重. 此外,Ni元素掺杂削弱程度最低. 3)电子结构计算分析了合金元素对Ag/Al界面稳定性影响的机制,掺杂使界面稳定性降低的主要原因是跨界面Ag-Al间的共价键成键作用减弱. 对Ca而言,Ca掺杂使界面处Al(1)-Ag(1)、Al(2)-Ag(2)的平均键布居值降低最严重,减小为0.193,0.188,对界面稳定性削弱程度最明显.对Ni而言,掺杂虽使Al(1)-Ag(1)的平均键布居值降低,但Al-Ni间更强的共价键作用削弱了该影响,使得Ni掺杂对界面稳定性的影响不明显.
4 结 论
猜你喜欢
东北水利水电(2022年6期)2022-06-28
科学技术创新(2022年15期)2022-05-18
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生理科应试(2021年10期)2021-12-07
中学生数理化(高中版.高二数学)(2020年6期)2020-07-24
电子制作(2019年11期)2019-07-04
小天使·五年级语数英综合(2015年4期)2015-04-20
中学化学(2014年6期)2014-09-09
