
硼烯/石墨烯异质结缺陷态电子结构的第一性原理研究
2022-12-07 09:36陈文浩刘会霞孙启花段海明
原子与分子物理学报 2022年1期
陈文浩, 马 洁, 刘会霞, 孙启花, 段海明
(新疆大学 物理科学与技术学院, 乌鲁木齐 830046)
1 引 言
自石墨烯成功分离[1]以来,二维材料就引起了人们极大的研究兴趣,如单层硅烯、锗烯、黑磷等[2-4],它们都有着各自独特的性质和特殊的用途. 硼烯作为最新的二维材料之一,2015年由美国阿贡国家实验室课题组[5]和中国科学院物理研究所课题组[6]先后独立成功合成,在机械性能上表现出比石墨烯更高的强度和柔韧性[7],在电学性能上,由于其各向异性的原子结构,硼烯表现出高度各向异性的金属性能[8]. 此外,硼烯的化学性质相对稳定,有助于克服硅烯和锗烯等易被氧化的缺点,在纳米电子器件方面具有良好的的应用潜质.
近年来,由两种或多种二维纳米薄层组装形成的二维异质结构(异质结)展现出诸多不同于单质二维纳米薄片的新奇物理性质和现象[9,10],基于异质结材料的器件具有传统器件所不具备的超薄、柔性及透明等特点,在新型微纳米光电子器件方面展现出有良好的应用前景[11,12]. 近年来,许多基于石墨烯的二维异质结(如磷烯、InSe以及MS2等与石墨烯相结合[13-16])被广泛研究. 最近,石墨烯-硼烯异质结也引起了研究者的关注:2019年首次在实验上成功制备了硼烯/石墨烯异质结结构[17],2020年A. Kochaev等研究了堆叠在单层石墨烯上的单层条状硼烯异质结结构,作者通过声子谱计算验证了结构的稳定性,能带结构计算表征了体系的金属性能[18].
在材料的合成制备中,通常都会存在几何结构缺陷,缺陷(态)会显著影响材料的力学、热学和电学性能[19]. Dongjing Liu用分子动力学方法研究了不同缺陷率石墨烯/硅异质结构的热导率[20],发现随缺陷率增加异质结热导率发生明显改变. Wei等人设计了一种平面石墨烯/六方氮化硼异质结构,采用分子动力学方法研究发现缺陷态的尺寸和位置对异质结力学性能有较大影响[21]. 然而,有关于硼烯/石墨烯异质结缺陷态的尺寸和位置对异质结电子性能的研究尚未报道. 因此,本文研究了缺陷态对单层硼烯与单质石墨烯相互作用所形成的二维异质结(single-layer striped borophene stacked on top of graphene,sB-Gr)的电子结构的调控作用,能够为基于含缺陷态的二维硼烯/石墨烯异质结的实验研究提供一定的理论参考.
2 计算方法
本文计算硼烯/石墨烯异质结采用了一个3×3×1的超胞来模拟异质结片状结构,如图1所示,图中不同的硼原子缺陷位点由数字1、2、3标识. 为避免相邻超胞沿层间的相互作用、沿超胞面外垂直方向保留20 Å的真空层. 具体计算采用VASP(ViennaAbinitiosimulation package)软件包[22],采取GGA-PBE交换关联势[23],使用平面波赝势基组,截断能设置为500 eV,布里渊区网格密度取7×7×1,几何结构弛豫中原子受力收敛标准为0.01 eV/Å、能量收敛标准为10-6eV,考虑异质结中层间相互作用计算中使用了DFT-D2范德华力修正[24].从头算分子动力学模拟采用NVT系综,所有计算均采用自旋非限制方式进行.
对于完整硼烯/石墨烯异质结(如图1所示),计算所得稳定(静态)结构中层间两侧的原子之间的最小距离为1.72 Å,这与A. Kochaev文章[18]计算结果完全一致,计算完整硼烯/石墨烯异质结3×3×1超胞的稳定(静态)结构能量为为-322.15 eV. 在分析缺陷态时考虑异质结中石墨烯一侧及硼烯一侧各自单独存在缺陷态的情形. 由于在硼烯/石墨烯异质结单胞中只包含相对独立的2个碳原子和3个硼原子,为充分考虑缺陷存在的可能性,对于单原子缺陷,对超胞中不等价位的两个碳原子分别取缺陷.当取石墨烯中最靠近硼烯的单个碳原子空位缺陷态时,经过直接的几何结构驰豫即发现空位对应的最近邻硼原子会脱离硼烯移动到石墨烯一侧,原有的硼烯/石墨烯层状异质结结构遭到破坏. 当单个碳原子空位缺陷态出现在距离硼烯次近邻位置时,可以发现其动力学结构也不再能表征层状异质结结构. 可见,对于硼烯/石墨烯异质结,石墨烯基底的完整性及硼烯与石墨烯基底间的相对弱的相互作用是异质结结构稳定存在的前提,在整个异质结中石墨烯在影响硼烯性能的同时起到支撑硼烯结构完整性的支撑作用,缺陷态只能出现在硼烯一侧.
3 结果与讨论
3.1 缺陷取位和缺陷密度
对于异质结中硼烯一侧的缺陷态,分别考虑三个不等价位硼原子的空位缺陷. 可以距离石墨烯一侧远近不同来标记不同类型硼原子:最远硼原子为1号位、次远硼原子为2号位、最近硼原子为3号位. 图1中分别标出了不同类型(1、2、3号位)的硼原子缺陷位点, 当单原子空位缺陷态出现在1号位硼原子处时,硼烯/石墨烯异质结3×3×1超胞的稳定(静态)结构能量为为-316.36 eV,当单原子空位缺陷态分别出现在2号位和3号位时,对应的静态结构能量分别为-315.87 eV和-317.42 eV. 体系相对更低的能量使得硼单原子空位缺陷态更易在3号位形成,此时体系(异质结)结构如图2所示.

图2 含单缺陷态硼烯/石墨烯异质结结构示意图. 左侧为顶视图,右侧为侧视图
考虑陷态密度增加至存在双原子空位缺陷情形. 此时,由单原子缺陷态构造双原子缺陷态存在多种取位方式. 对这些可能的双空位缺陷结构进行系统计算,可发现不同情形:对于1号位-2号位、及1号位-3号位硼原子空位形成的双原子缺陷,经过结构驰豫和分子动力学模拟后均发现在硼烯一侧围绕该类双空位缺陷硼原子发生团聚化形成小团簇,从而不再表征二位异质结结构.
当双原子空位缺陷出现在2号位-3号位时,体系能量较高,相较于其他几种缺陷态结构不具有能量优势. 由于单原子空位缺陷态时3号位缺陷态的相对明显高的稳定性(相较1号位及2号位缺陷态具有明显的能量优势),考虑双原子空位缺陷均出现在3号硼原子空位的情形,此时、由于硼原子空位缺陷的排布不同会出现两种双原子空位缺陷态:接触式双原子空位缺陷态和非接触式双原子空位缺陷态,图3中给出了这两种不同缺陷态的结构示意图.结构弛豫及常温分子动力学模拟表明两种缺陷态结构均能够稳定存在(二维异质结结构能够保持),但相较非接触式缺陷态结构(能量为-311.29 eV)、接触式缺陷态结构具有更高的稳定性(对应于更低的能量-315.76 eV).
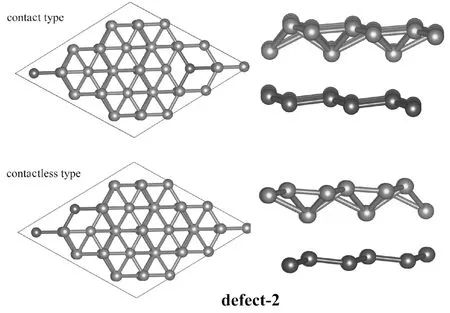
图3 含双缺陷态硼烯/石墨烯异质结结构示意图. 左侧为顶视图,右侧为侧视图,上图为接触式,下图为非接触式
当缺陷态密度增加至三空位缺陷时,从能量和结构稳定性两方面进行考虑,三个空位缺陷均出现在3号位硼原子处时具有明显的竞争优势.由于缺陷位置在硼烯中的不同排布,会出现四种三缺陷态结构模型,如图4所示. 当缺陷空位各不相邻,可将其命名为缺陷非接触式结构(contactless-type),相对应静态结构能量为-309.31 eV;当各缺陷空位均为近邻排布时,可将其命名为聚拢式结构(gather-type),相对应静态结构能量为-308.72 eV;当缺陷空位呈链状排布时称其为串联式线状结构(line-type),静态结构能量为-308.43 eV;当缺陷空位相互近邻并呈拐角式排布时将其命名为拐角式结构(corner-type),静态能量为-310.41 eV.可见,拐角式三空位缺陷态结构具有更低的能量和更高的结构稳定性,在形成过程中更具有能量优势.

图4 含三缺陷态硼烯/石墨烯异质结结构示意图. 左侧为顶视图,右侧为侧视图. 自上而下分别对应于聚拢式结构、非接触式结构、拐角式结构、线状结构
3.2 分子动力学稳定性
采用分子动力学模拟研究含缺陷态硼烯/石墨烯异质结的动力学稳定性. 模拟均采用恒温分子动力学方法(NVT系综). 模拟中温度取为300 K、时间步长为1 fs、总模拟时长为3 ps. 以如上含(单、双、三)缺陷态时能量最低的静态结构(称之为理想结构)为初始结构. 图5(a)、(c)及(e)分别对应于初始结构为包含单、双、三缺陷态理想结构硼烯/石墨烯异质结的温度-时间及能量-时间变化曲线.
分析图5可见,对于含单原子缺陷态异质结,从理想结构出发,在起始过程中会出现能量的明显降低,这表明含单缺陷态的初始理想异质结结构处于亚稳定. 由图5(a)可见,到500步以后能量曲线呈现振荡式变化、振荡幅度变小、能量变化渐趋稳定. 可以对500步之后的任一个动力学结构进行几何结构弛豫、其对应的稳定静态结构(可称之为淬火结构)属性基本相同(能量及能带结构基本一致),并且均能够表征异质结特征. 以该淬火结构为初始结构,对包含单缺陷态的异质结再进行同样(温度为300 K、时间步长为1 fs、总模拟时长为3 ps)的分子动力学模拟,所得结果(温度-时间及能量-时间变化曲线)示于图5(b)中,此时,能量变化整体上无平移现象、只是围绕在一个恒定值上下波动,表明该淬火结构为动力学稳定结构. 可见,包含单缺陷态的硼烯/石墨烯异质结可以稳定存在.
对于含双缺陷态的硼烯/石墨烯异质结的恒温分子动力学模拟结果与如上含单缺陷态异质结结果类似. 从理想结构出发,在起始过程中(约前300步)会出现能量的明显降低,这表明含双缺陷态的初始理想异质结结构处于亚稳定.
由图5(c)可见,到300步以后能量曲线呈现振荡式变化、变化趋势渐趋稳定. 对300步之后的多个不同动力学结构进行几何结构弛豫、所得的稳定静态结构(淬火结构)属性(能量及能带结构)也基本相同,并且也均能够表征异质结特征. 同样,以该淬火结构为初始结构,对包含双缺陷态的异质结也再进行同样的分子动力学模拟,所得结果(温度-时间及能量-时间变化曲线)示于图5(d)中,此时,能量变化整体上也无平移现象、表现为围绕某个恒定值的上下波动,表明该淬火结构也是动力学稳定结构. 可见,包含双缺陷态的硼烯/石墨烯异质结也可以稳定存在.

图5 (a)、(b) 含单缺陷态硼烯/石墨烯异质结的温度及能量随时间的变化((a)图初始结构为理想结构、(b)图初始结构为淬火结构).(c)、(d)含双缺陷态硼烯/石墨烯异质结的温度及能量随时间的变化((c)图初始结构为理想结构、(d)图初始结构为淬火结构).(e)含三缺陷态硼烯/石墨烯异质结的温度及能量随时间的变化(初始结构为理想结构)
对于包含三缺陷态的硼烯/石墨烯异质结的恒温分子动力学模拟结果与如上含单、双缺陷态异质结结果不同. 从三缺陷态理想结构(拐角式三空位缺陷态结构,参见图4)出发,在整个模拟时间段中,体系能量曲线一直呈现振荡式变化,表明该初始结构是动力学稳定的.可见,包含三缺陷态的拐角式三空位缺陷态硼烯/石墨烯异质结可以稳定存在.
3.3 缺陷态结构的电学性能
体系的物理化学性质会受到缺陷态的影响.因缺陷态的存在会导致围绕缺陷态的电子分布与完整(无缺陷态)结构明显不同,从而会改变体系的电子结构,可能会引起体系导电性能的改变. 图6中给出了完整硼烯/石墨烯异质结及分别包含单、双、三缺陷态最低能量稳定结构硼烯/石墨烯异质结的能带结构与分波态密度图.
无缺陷完整硼烯/石墨烯异质结的能带结构如图6(a)所示,有多条能带穿过费米能级,呈现出无能隙的金属特性,并且为非磁性体系. 分析图6(b)分波态密度可见,在费米能级附近体系电子态主要由碳的p轨道电子贡献,其次是硼的p轨道电子贡献次之,硼的s轨道电子贡献比碳的s轨道贡献明显要大,但总体上费米能处s电子贡献远小于p电子,体现体系导电性能的电子基本由p电子贡献.
图6(c)、(e)分别给出了含单、双缺陷态硼烯/石墨烯异质结的能带图,图6(d)、(f)分别给出了含单、双缺陷态硼烯/石墨烯异质结的分波态密度图. 同无缺陷态的完整异质结类似,在包含单、双缺陷态硼烯/石墨烯异质结的能带图中,在其费米能处均有能带穿越,均体现出导电的金属特性. 同样,分析二者分波态密度图,也可以发现在费米能处电子主要由C、B原子的p电子贡献(C的p电子贡献仍旧多于B的p电子).
图6(g)、(h)分别给出了含三缺陷态硼烯/石墨烯异质结的能带图及分波态密度图.

图6 (a)、(b)完整硼烯/石墨烯异质结的能带与分波态密度.(c)、(d)含单缺陷态硼烯/石墨烯异质结的能带与分波态密度.(e)、(f)含双缺陷态硼烯/石墨烯异质结的能带与分波态密度.(g)、(h)含三缺陷态硼烯/石墨烯异质结的能带与分波态密度
与完整异质结及含单、双缺陷态异质结所体现出的金属性显著不同的是,在含三缺陷态硼烯/石墨烯异质结的能带图中费米能处无能带穿过、体系存在能隙,具体表现为具有约0.43 eV带隙的间接带隙半导体特性.
此外,分析含三缺陷态异质结分波态密度(图6(h))可见,其靠近费米能的占据态(非占据态)主要由B原子的p电子贡献(强于C原子的p电子贡献),这与完整异质结及含单、双缺陷态异质结所体现出的费米能处电子态处主要来源于C的p电子的特征也明显不同.
可见,对于硼烯/石墨烯异质结,伴随缺陷态的出现和缺陷态密度的增加,异质结会发生导电性能的转变:从完整结构或缺陷态密度较低(含单、双缺陷态)时的金属性转变为缺陷态密度较高(含三缺陷态)时的半导体特性.
4 结 论
本文采用基于密度泛函理论的第一性原理计算方法系统研究了缺陷态位置和密度对硼烯-石墨烯异质结的结构稳定性及导电性能的影响. 结果表明,当缺陷位点出现在石墨烯一侧时将会导致二维双层硼烯-石墨烯异质结结构的可辨识度消失,异质结结构遭到破坏,因此在确保硼烯-石墨烯异质结结构特性稳定存在的前提下异质结中空位缺陷态只能出现在硼烯一侧. 通过分析体系的能带图及分波态密度图可以发现:完好结构的硼烯-石墨烯异质结及含单、双缺陷态的硼烯-石墨烯异质结均为导体,且其导电电子主要由C、B原子的p电子贡献;而含三缺陷态的硼烯-石墨烯异质结为间接带隙半导体,带隙为0.43 eV. 这表明对于硼烯-石墨烯异质结、可以通过调控体系的缺陷态分布及数目(密度)来调控体系的电子结构,从而实现导体到半导体的转变.
猜你喜欢
物理学报(2022年17期)2022-09-14
小天使·聪聪画刊(2021年2期)2021-09-10
汽车零部件(2020年10期)2020-11-09
汉语世界(The World of Chinese)(2019年6期)2019-09-10
体育世界(学术版)(2019年4期)2019-06-13
陶瓷学报(2019年5期)2019-01-12
汽车生活(2018年5期)2018-06-21
小学生·多元智能大王(2015年4期)2015-05-18
读者欣赏(2014年6期)2014-07-03
青年文摘·上半月(1984年3期)1984-11-01
