
红树植物拟海桑及其亲本的根际细菌群落特征分析
2022-12-05 12:53:54叶锦成陈毅青高琳周鲜娇钟才荣张颖王芸
热带海洋学报 2022年6期
叶锦成, 陈毅青, 高琳, 周鲜娇, 钟才荣, 张颖, 王芸
1. 岭南师范学院红树林研究院, 广东 湛江 524048;
2. 暨南大学生命科学技术学院生态学系, 广东 广州 510632;
3. 海南省林业科学研究院(海南省红树林研究院), 海南 海口 571129
红树林湿地生态系统广泛分布于热带、亚热带海岸, 具有美化环境、保护海岸、支持沿岸区域水生生物的栖息和觅食等生态服务功能(Giri et al,2011; Alongi, 2014; 江睿 等, 2021)。红树林湿地还拥有丰富的生物多样性, 但由于气候变化、海平面上升和人为活动的影响, 红树林湿地面积迅速减少,全球每年会减少1%~2%(Duke et al, 2007; Giri et al,2011; Luis et al, 2019)。在过去的半个世纪里, 红树林湿地的面积已经下降了 30%~50%(Duke et al,2007; Polidoro et al, 2010; Donato et al, 2011)。如何保护并修复红树林湿地生态系统是当前红树林生态保护工作的一个重要研究内容。
海桑属(Sonneratia)红树植物是红树林的重要树种之一, 通常被认为是红树林海岸的先锋种(毛礼米等, 2009; 李诗川 等, 2014), 开展海桑属红树植物的研究对红树林生态系统的修复具有重要意义。在我国的海桑属目前包含 6 个有效种(Duke, 1994;Zhong et al, 2020), 分别为: 海桑(S.caseolaris)、杯萼海桑(S.alba)、卵叶海桑(S.ovata)、拟海桑(S.×gulngai)、海南海桑(S.×hainanensis)和无瓣海桑(S.apetala)。拟海桑(S.×gulngai)最早由Muller 等(1966)发现, 但未正式命名, 后由Duke(1984)于澳大利亚东北部再次发现并命名。1993 年, 华南植物所高蕴璋(1993)在我国海南岛首次发现海桑属新种拟海桑。王瑞江 等(1999)通过形态学、花粉学、细胞学以及其他方面的比较研究, 认为在中国海南发现的拟海桑为杂交种, 它的嫌疑亲本为杯萼海桑(S.alba)和海桑(S.caseolaris), 并归并入S.×gulngaiN. C. Duke。植物分类学家通过对拟海桑的分类学特征研究发现, 拟海桑是海桑和杯萼海桑的自然杂交种, 拟海桑及其亲本是广布种, 在中国海南省以及澳大利亚等地均有分布(Muller et al,1966; 高蕴璋, 1993; 王瑞江 等, 1999; Zhong et al,2020)。王瑞江 等(1999)通过对野外杂交种植株的形态学观察发现, 拟海桑植株个体明显比杯萼海桑和海桑高大, 这与Duke(1984)对澳大利亚的海桑植物杂种研究结果一致。
已有大量的研究表明, 根际微生物在植物的生长发育和植物病虫害的生物防治等方面都具有十分重要的生态学意义(李晴晴 等, 2019; 刘京伟 等,2021)。此外, 宿主遗传学和植物微生物组成之间存在相互作用, 其中与根相关的微生物群落部分会受宿主遗传因素的影响(Peiffer et al, 2013; Schlaeppi et al, 2014; Edwards et al, 2015; Naylor et al, 2017;Deng et al, 2021), 植物表型能作为微生物组功能的预测因子和读数(Wagner, 2021)。最新研究表明, 根际微生物群还可以促进植物表型可塑性, 可以通过植物激素的产生来调节植物的生长发育(Lu et al,2018)。Wang 等(2010)还从红树植物中分离出一种在温室被证实能够显著改善实验植物生长的AMF(arbuscular mycorrhizal fungi), 可导致植物的高度、地面直径和生物量增加, 并使实验植物对N、P 和K 的吸收增加。因此研究子代杂交种拟海桑与其亲本的根际微生物群落关系对于解释杂交种拟海桑生长优势具有重要意义。
尽管目前研究者们已经在植物微生物群、根系分泌物和植物生长繁殖之间的相互作用等方面取得了重大进展, 但是对海桑属红树植物根际微生物这方面的报道仍然较少(Yang et al, 2014; 颜栋美 等,2018; Muwawa et al, 2021)。针对野外杂交种拟海桑生长能力较强的特性, 国内外尚没有相关文献对造成这种现象的原因进行解释。目前第二代测序技术——高通量测序技术因其高效性、准确性成为研究土壤微生物的主要方法(Margulies et al, 2005;Quail et al, 2008; 周晓光 等, 2010; 闫绍鹏 等,2012)。故本研究基于高通量测序技术对中国海南岛东寨港的杂交种拟海桑及其亲本海桑和杯萼海桑的根际细菌的群落结构特征进行研究, 分析杂交子代与亲本间的根际细菌菌群结构特征的差异, 对解释拟海桑的生存优势和红树林湿地生态系统的保护和修复工作具有参考价值。
1 材料与方法
1.1 实验材料
本实验样本于2018 年6 月采集自海南省东寨港红树林保护区内, 在采样区100m×200m 的区域中,采用蛇形采样, 每种植物每隔30m 选一个点, 利用土壤取样器在植物根区, 分别采集5 份土壤混合均匀后装入无菌样品袋中, 并保存在专门的便携式冷却器中, 共获得土样9 个。采集当天运回实验室进一步处理, 在实验室中, 每个土样被分成两份: 一份用于土壤理化性质分析, 在4℃下储存, 另一份在–80℃下储存用于进一步的根际土壤总DNA 提取。本研究所采样本海桑的缩写为Sc, 杯萼海桑缩写为Sal、拟海桑缩写为Sg。
1.2 土壤化学性质测定
土壤全氮(total nitrogen, TN)和土壤全碳(total carbon, TC)利用碳氮元素分析仪(Elementar Vario EL Ⅲ, Elementar, 德国)测定(张威 等, 2009)。土壤全磷(total phosphorus, TP)采用钼锑抗显比色法测出,速效钾(available potassium, AK)采用醋酸铵浸提—火焰光度计法测出(鲍士旦, 2000), 每个土样重复3 次。
1.3 土壤细菌群落组成测定
1.4 生物信息学分析
1.4.1 Alpha 多样性分析
单样本的OTU 多样性分析(alpha diversity)可以反映样本内微生物群落的丰富度和多样性, 利用每个OTU 在样品中的有效序列的绝对丰度和相对信息(Edgar, 2013), 使用 Qiime(Version1.9.1)计算Observed-OTUs、ACE、Shannon、Simpson 和Chao1等指数, 以评估各样本中微生物群落的物种丰富度和多样性等差异。Chao1 和ACE 指数简单指群落中物种的数量, 而不考虑群落中每个物种的丰度情况。Shannon 指数和Simpson 指数受样品群落中物种丰度和物种均匀度的影响。
1.4.2 Beta 多样性分析
利用Qiime(Version1.9.1)软件对所测所有样本的细菌群落构成进行分析, 采用主坐标分析(PCoA)和PERMANOVA 对样本间的细菌种群结构差异显著性进行分析。
1.4.3 细菌类群的分类
为更好地寻找影响微生物群落的关键种, 确定所有微生物类群的作用和贡献, 根据Dai 等(2016)提出的物种丰度划分, 把稀有类群的阈值设置为0.1%, 丰富类群的阈值设置为1%, 并根据它们的丰度将所有可操作的分类单元分为如下6 个专属类别:稀有物种(rare taxa, RT), 即在所有的样本中丰度均低于0.1%; 丰富物种(abundant taxa, AT), 在所有的样本中丰度均高于1%; 中等物种(moderate taxa,MT), 在所有样本中丰度在0.1%至1%之间; 条件稀有物种(conditionally rare taxa, CRT), 在所有样本中丰度均低于1%, 同时在部分样本中丰度低于0.1%;条件丰富物种(conditionally abundant taxa, CAT), 在所有样本中丰度均高于0.1%, 同时在部分样本中丰度高于1%; 条件稀有或丰富物种(conditionally rare or abundant taxa, CRAT), 在部分样本中的丰度在低于0.1%, 同时在部分样本中的丰度高于1%。使用R语言(Version3.5.0)按照拆分标准把所有的OTUs 进行拆分。
1.4.4 PICRUSt 功能预测
PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states)功能预测是基于微生物16S 序列间的进化距离以及已知全序列的微生物的功能基因拷贝数, 对其他微生物物种功能基因的拷贝数进行推测, 进而估计整个样品中各个功能基因的丰度分布的一种方法。其可以较准确地对微生物群落的功能进行分析。根据PICRUSt 开发者的评测, 对于NSTI<0.05 的肠道微生物样品和NSTI≈0.17 的土壤样品, PICRUSt 的预测结果较好, 而对于 NSTI≈0.23 的微生物样品,PICRUSt 的预测结果较差。该方法首先通过祖源序列重建算法, 对GreenGene 数据库中收录的微生物进行16S rRNA 拷贝数和KEGG(Kyoto encyclopedia of genes and genomes) 、 eggCOG(Evolutionary Genealogy of Genes: Clusters of Orthologous Groups of proteins)及RFAM 功能注释对应基因的拷贝数进行推测, 得到每个物种单位拷贝的16S rDNA 对应的功能注释的丰度。然后根据上一步推测得到的数据, 对样品中各个物种的16S rRNA 拷贝数进行校正。之后根据校正后的16S rRNA 拷贝数, 对样品中各个功能或注释条目的拷贝数进行估计, 并进行后续的差异分析。最后根据KEGG 和eggCOG 数据库中的分层分类信息, 在不同层级将功能基因进行归类, 并计算不同分层水平下, 不同类别功能基因在样品中的丰度(Langille et al, 2013)。
现代企业的管理者和领导者要想继续提升自身的能力和技能,大多通过商学院教育获得,途径相对单一。目前,虽然商学院的发展已经初具规模,但是远远不能满足当前企业的需要,这也是加强商学院人才培养,积极拓展路径的现实需要。商学院的发展必然跟经济体系的发展有着直接的关系,作为经济生活的重要参与者和建构者,商学院的发展与经济发展密切相关,商学院的发展要顺应经济发展的需要和趋势,在人才培养路径上要切合经济市场的需求。
1.5 数据统计
使用Qiime(Version1.9.1)软件评估样品的根际土壤细菌菌群多样性, 计算多样性指数后使用SPSS(Version13.0)进行ANOVA+Turkey HSD test。根据不同的距离矩阵计算方式计算 Unweighted Unifrac, Weighted Unifrac, Bray Curtis, Euclidean 距离矩阵后使用R 语言(Version 3.5.0)进行PCoA 分析和作PCoA 图进行Beta 多样性展示(Hamady et al,2010; Yatsunenko et al, 2012; Jiang et al, 2013)。使用Microsoft Excel 2016 对测序所得的原始OTUtable中的各分类菌群进行丰度的排序, 选择优势菌群使用GraphPad Prism(Version 8.0)绘制相对丰度累加图展示。此外还使用SPSS(Version13.0)对土壤理化性质环境因子进行ANOVA+Duncan 检验以及其和菌群的相关性分析(Spearman), 检验显著性水平为P<0.05。此外PERMANOVA 组间相似性统计分析、物种丰度拆分、热图、三元图的绘制均由R 语言(Version 3.5.0)进行(Oksanen et al, 2007)。
2 结果与分析
2.1 测序数据
对三种样品, 每种三个重复, 共9 个样本进行测序, 测序结果如表1。根据测序数量可知, 每个样本的测序数量均超过3 万条, 稀释性曲线趋于平台期(数据没有显示), 说明结果能够比较全面真实地反映根际土壤微生物群落组成。聚类后共获得11475个有效OTU(97%的相似度)。共获得30 门, 242 科,251 属的根际土壤细菌。门、科、属、种的可注释水平分别为86.95%、41.53%、10.41%和0.36%。
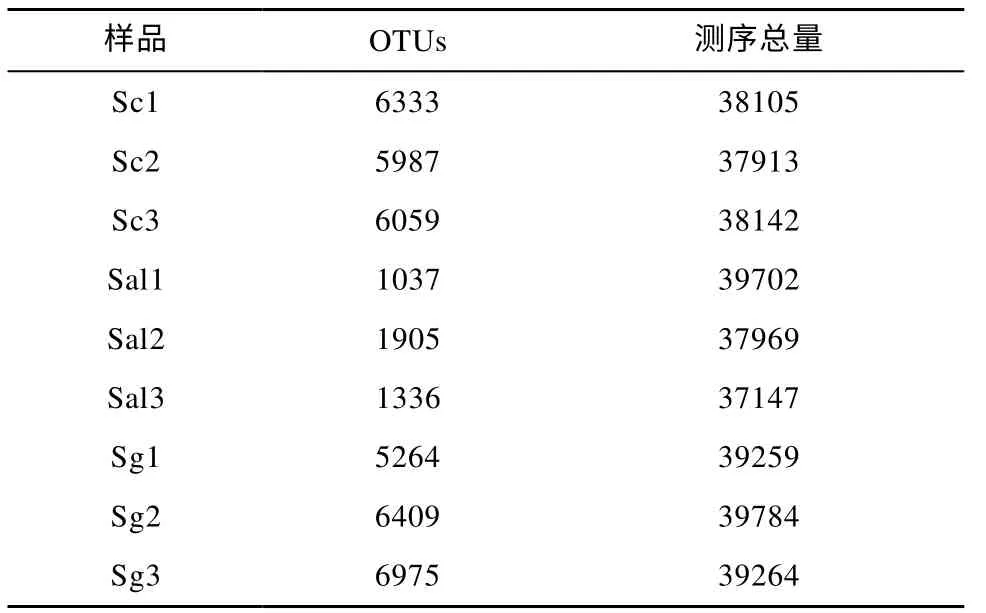
表1 土壤样本测序数据量统计Tab. 1 Statistics of soil samples volume sequencing data
2.2 细菌群落结构组成与差异性分析
在门水平上, 由图1a 可知, 三种样品的根际土壤细菌主要类群包括变形菌门(Proteobacteria)、绿弯菌门(Chloroflexi)、浮霉菌门(Planctomycetes)、拟杆菌门(Bacteroidetes)、酸杆菌门(Acidobacteria)、栖热菌门(Thermi)和厚壁菌门(Firmicutes)7 个类群。变形菌门是所有样本共同的绝对优势门, 在各组样本中占比均达到40%以上。不同样品的丰富细菌类群种类和相对丰度具有差异。在Sc 样品中丰度排名前四的门依次是变形菌门(Proteobacteria) 40.38%、绿弯菌门(Chloroflexi)14.37%、浮霉菌门(Planctomycetes)6.02%、拟杆菌门(Bacteroidetes)4.33%。丰度占比达65.15%。在Sg 样品中丰度排名前四的门依次是变形菌 门 (Proteobacteria)45.78% 、 浮 霉 菌 门(Planctomycetes)11.25% 、 绿 弯 菌 门(Chloroflexi)6.84%、酸杆菌门(Acidobacteria)6.5%。丰度占比达70.37%。在Sal 样品中丰度排名前四的门依次是变形菌门(Proteobacteria)47.35%、栖热菌门(Thermi)19.84%、拟杆菌门(Bacteroidetes)13.32%、厚壁菌门(Firmicutes)5.86%。丰度占比达86.37%。其中Sal样品的丰度丰富细菌类群在三种样品中丰度占比最高, 具有高优势。从丰度丰富细菌类群组成方面分析, 样品Sg 的菌群各门与Sc 更为相似, 样品Sal 的菌群组成与Sc、Sg 的差异较大。从丰度方面分析, 丰富细菌类群在子代和亲本的丰度具有显著差异(图2),例如 Sg 的酸杆菌门(Acidobacteria)和放线菌门(Actinobacteria)的丰度分别为4.3%和6.5%, 显著高于亲本的1%~2%的含量, 且与Sc 比较更为显著。在Sal中占比高达19.8%的栖热菌门(Thermi)在Sg 和Sc 中占比少于1%。占比5%的热袍菌门(Thermotogae)在Sg 中消失。在属水平上, 结合图1b 分析, 发现在属水平上不同样品间的丰度丰富根际细菌类群的组成也具有较大的差异。在数量上, 随着分类水平的降低, 三种样品属水平丰度丰富菌群种类减少。此外Sg 浮霉菌门的Planctomyces属相对丰度高于其他样品, 栖热菌门的g__B-42 属和变形菌门的Sulfurimonas属的相对丰度低于其他样品。
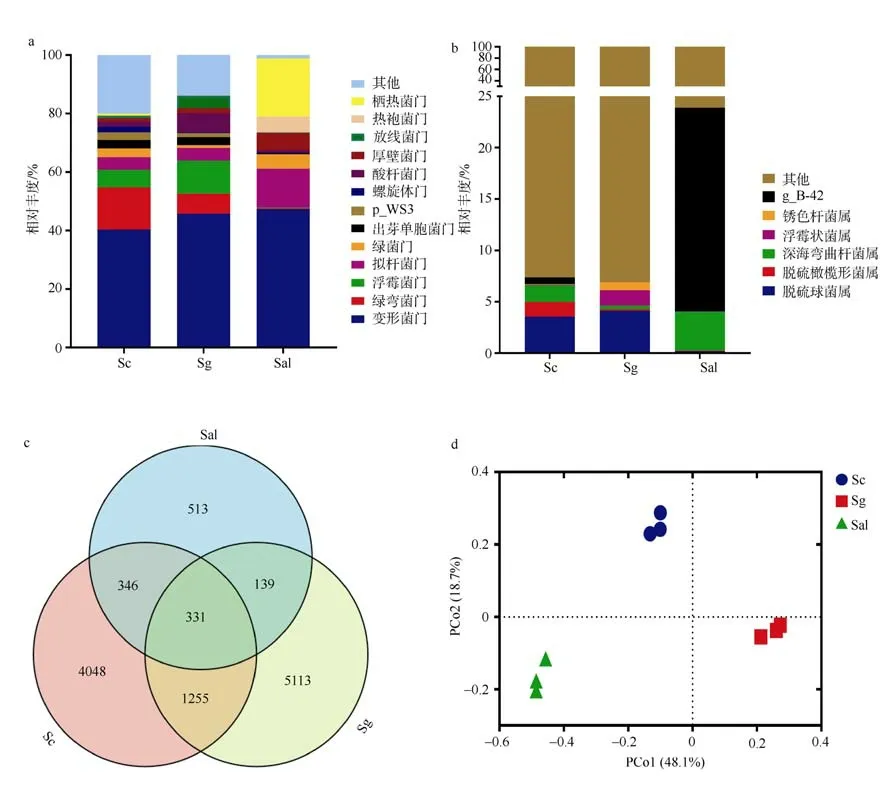
图1 各样本细菌群落构成与比较。a. 各样品菌群组成在门水平相对丰度; b. 各样品菌群组成在属水平相对丰度; c. Venn 图展示不同样本的共有和特有的OTU 数量; d.基于Weighted unifrac 距离的主坐标分析Fig. 1 Composition and comparison of bacterial community in each mangrove species. (a) Relative abundance map at phylum level. (b) Relative abundance map at genus level. (c) The Venn diagram shows the number of common and unique OTUs for different samples. (d) Principal coordinate analysis generated using Weighted unifrac distance
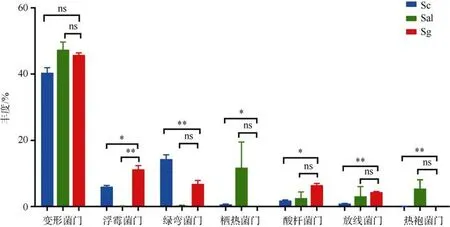
图2 主要细菌菌群(门水平)的丰度差异配对样本T 检验, ***P<0.001; **P<0.01; *P <0.05; ns P>0.05Fig. 2 The difference in the abundance of the main bacterial flora(Phylum level, Paired-Samples T Test , ***P<0.001;**P<0.01; *P <0.05; ns P>0.05)
2.3 Venn 图分析
通过维恩图可以直观地比较不同海桑样品根际土壤样本间共有和特有的OTU 数量, 并反映出样本在OTU 水平上的细菌物种组成差异(图1c)。三种海桑样品, 9 个样本共测出11475 条OTU, 其中Sc5980条、Sg6838 条、Sal1329 条。三种样品共有OTU 数目为331, 占比2.8%。Sc 和Sg 共有的细菌OTU 数量为1255, 同时Sc 特有OTU 数量为4048, Sg 为5113; Sc 和Sg 两者共有细菌OTU 数量占14.12%,同时Sc 和Sg 各自特有的OTU 数量分别占35%和43.5%。Sal 和Sg 共有的细菌OTU 数量为139, 同时Sal 特有OTU 数量为513, Sg 为5113; Sal 和Sg两者共有细菌OTU 数量占1.18%, 同时Sal 特有的OTU 数量占4.36%。说明三种样品中菌群结构差异程度各不相同。Sc 和Sg 的共有OTU 数目最多, 差异程度最小, 相似性最高。Sal 和Sg 的差异程度大,相似性低。
2.4 Alpha 多样性分析
根际细菌群落的各α多样性指数的计算结果表明, 三种海桑根际细菌群落多样性高(表2)。对样本间的多样性指数进行ANOVA+Turkey HSD test发现, Sc 和 Sg 样本的 Observed-OTUs、ACE、Shannon 指数、Simpson 指数和Chao1 指数数值相似性高, 而 Sal 样本的 Observed-OTUs、ACE、Shannon 指数、Simpson 指数和Chao1 指数与Sc和Sg 样本比较显著偏低。表明样本Sc 和样本Sg间的多样性无显著性差异, 而Sc 与Sal、Sal 与Sg之间存在显著差异性。
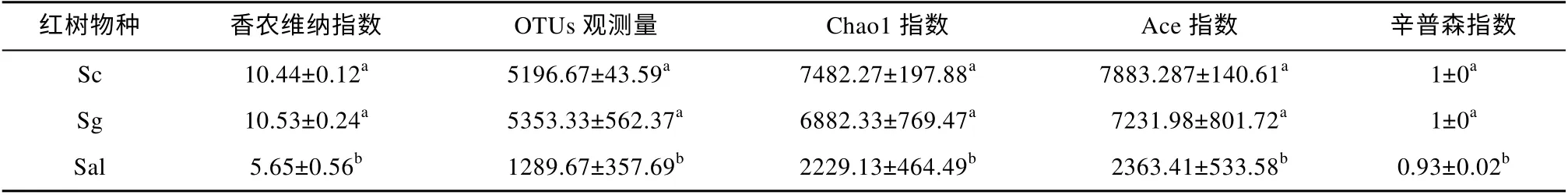
表2 三种海桑的根际细菌群落α多样性指数分析(平均值±标准误差)Tab. 2 Alpha diversity index of rhizosphere bacterial community of three species of Sonneratia (Mean±SD)
2.5 Beta 多样性分析
Beta 多样性统计分析揭示了样本之间的细菌群落差异, 基于Bary-Curtis、Enclidean、unweighted Unifrac、weighted Unifrac 距离。利用PCoA 分析, 不同距离矩阵PCoA 结果均表明不同海桑样品之间的根际微生物细菌的群落组成具有一定的差异性, 其中Weighted Unifrac 距离关注样品间物种种类以及数量的组成差异。不同样本间的Weighted unifrac distance 均值Sc 与Sg 为0.57, Sal 与Sg 为0.73, Sc与Sal 为0.73。PCoA 分析结果(图1d)显示Sg 与Sc的距离值更短, 表明它们根际细菌菌群的物种种类以及数量的组成更为相似。 进一步利用PERMANOVA 检验发现三种样品根际土壤细菌群落组成差异并不显著。
2.6 物种丰度划分
所有细菌OTU 被划分到6 个分类群类别, 对于潜在的区分群落的关键类群AT、CAT、CRAT 的归类情况如下: 没有OTU 被归类为AT(在所有样本中相对丰度都高于1%的OTU), CAT(仅在一些样本中相对丰度高于1%, 但从不低于0.01%的OTU)包含2个OTU, 均属于变形菌门, CRAT 组(在部分样本中的相对丰度低于0.1%, 同时在部分样本中的相对丰度高于1%)包含31 个OTU。研究认为微生物群落的特征是少数优势高丰度分类群和低丰度稀有分类群影响的结果(Sogin et al, 2006; Pedrós-Alió, 2007;Galand et al, 2009), 参考Dai 等(2016)的研究方法,对本研究的每组划分结果进行综合分析后认为本研究的CRAT 组最具代表意义。对该组的物种绘制热图(图3)直观地呈现出多个样本多个OTU 的全局丰度分布变化和多OTU 丰度分布的聚类关系, 并绘制三元图(图 4)分析各根际细菌群落之间的差异性OTUs 的贡献度, 等边三角形中圆点的位置由三种海桑属样本的根际微生物群对各OTU 相对丰度的贡献确定(Dombrowski et al, 2017)。由图3 可知,CRAT 组中Sc 样本的高丰度OTU 均属于变形菌门(2 个), Sg 样本的高丰度OTU 属于变形菌门(4 个)和厚壁菌门(2 个)。Sal 样本的高丰度OTU 属于变形菌门(12 个)、拟杆菌门(4 个)、绿菌门(3 个)、厚壁菌门(2 个)、热袍菌门(1 个)和栖热菌门(2 个)。三种样品根据多组值间两两的差异程度(欧氏距离)进行聚类发现样本在系统关系上(图上方的树状结构)分为了两类。一类是Sc 和Sg, 另一类是Sal。结合三种样本关键类群的数量和分类的差异说明了菌群的相似程度为Sc 和Sg 更相似, Sal 样本中的菌群异质性相对较高。图4 中三角形的三个点分别代表三个样品组, 从图中看出距离Sc 近的有一个OTU, 来自变形菌门, 距离Sg 近的有两个OTU 来自变形菌门和拟杆菌门, 距离Sal 近的也有两个OTU 来自厚壁菌门和变形菌门。综合图3 和图4 分析, 三种样品中确实存在能区分三种样品根际土壤细菌群落的关键OTU。
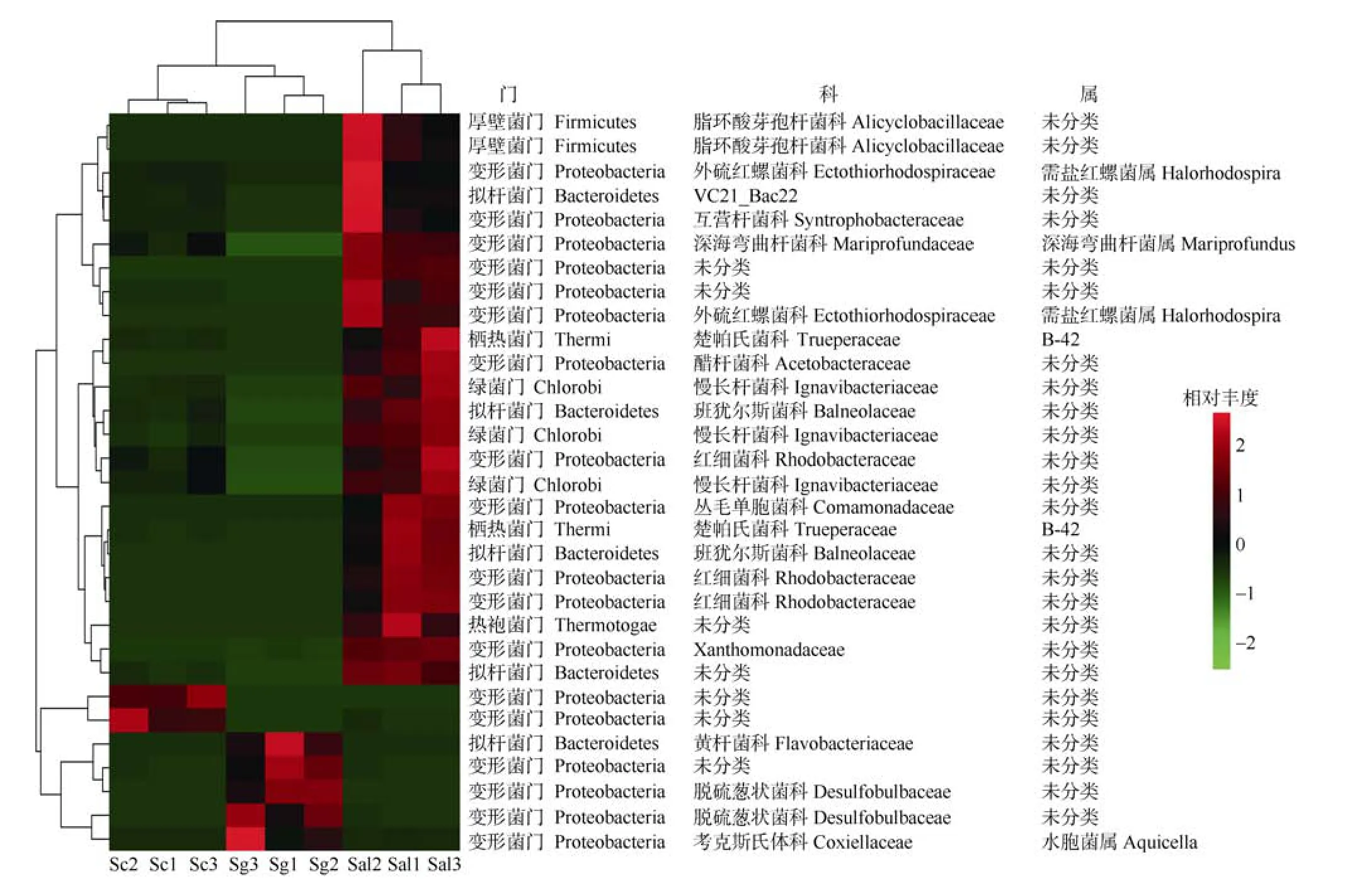
图3 各样本CRAT 拆分组OTU 水平热图Fig. 3 Heat map of OTU level of each sample CRAT split group
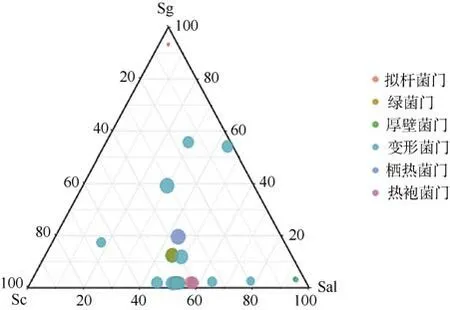
图4 各样本CRAT 拆分组OTU 水平的三元相图Fig. 4 The ternary plot of the OTU level of each sample CRAT split group
2.7 土壤理化性质与微生物群落多样性的关系
对不同样品土壤测定了全碳(TC)、全氮(TN)、全磷(TP)和速效钾(AK)含量, 并分析计算得出碳氮比(C/N)和氮磷比(N/P), 各数值如表3 所示。结果显示Sc 样品的土壤全碳含量较高; Sc 和Sal 的根际土壤全氮量比Sg 高3 倍以上。
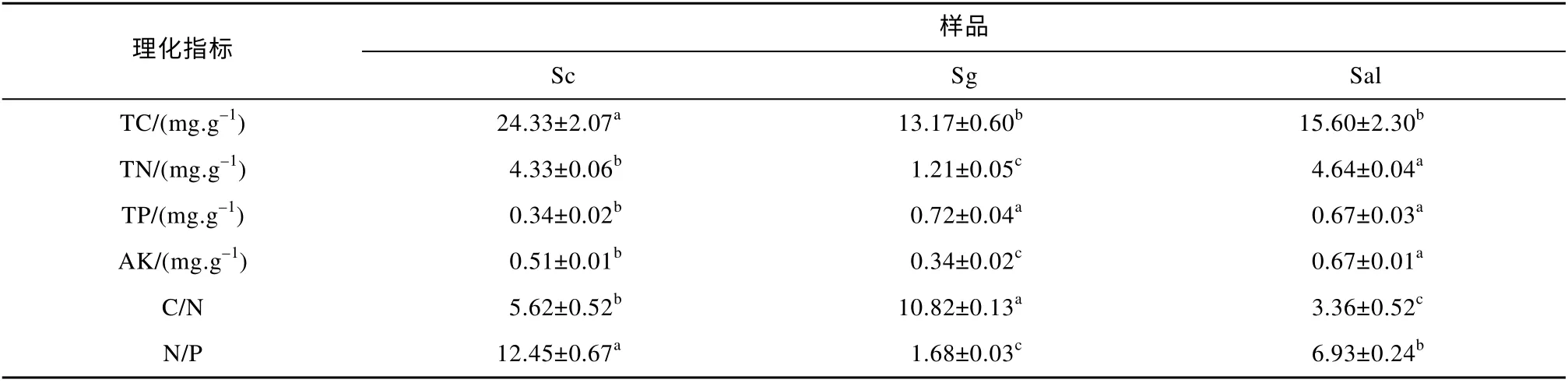
表3 土壤样品的理化性质(平均值±标准误差)和ANOVA 显著性分析、Duncan 检验多重比较结果Tab. 3 Physicochemical characteristics of soil samples (Mean±SD) and ANOVA significance analysis, Duncan test multiple comparison results
应用相关分析方法研究TC, TN, TP, AK, C/N,N/P 和变形菌门、绿弯菌门、浮霉菌门、拟杆菌门、绿菌门、芽单胞菌门、WS3、螺旋体门、酸杆菌门、厚壁菌门、放线菌门、栖热菌门、热袍菌门之间的相关关系, 使用Spearman 相关系数表示相关关系的强弱情况, 结果如表4 所示。由表4 可知, TC 与变形菌门丰度具有显著负相关关系; TN 和浮霉菌门、酸杆菌门、放线菌门丰度之间具有显著负相关关系,而与绿菌门、厚壁菌门、栖热菌门、热袍菌门丰度之间有着显著的正相关关系; TP 与变形菌门丰度具有显著正相关关系。
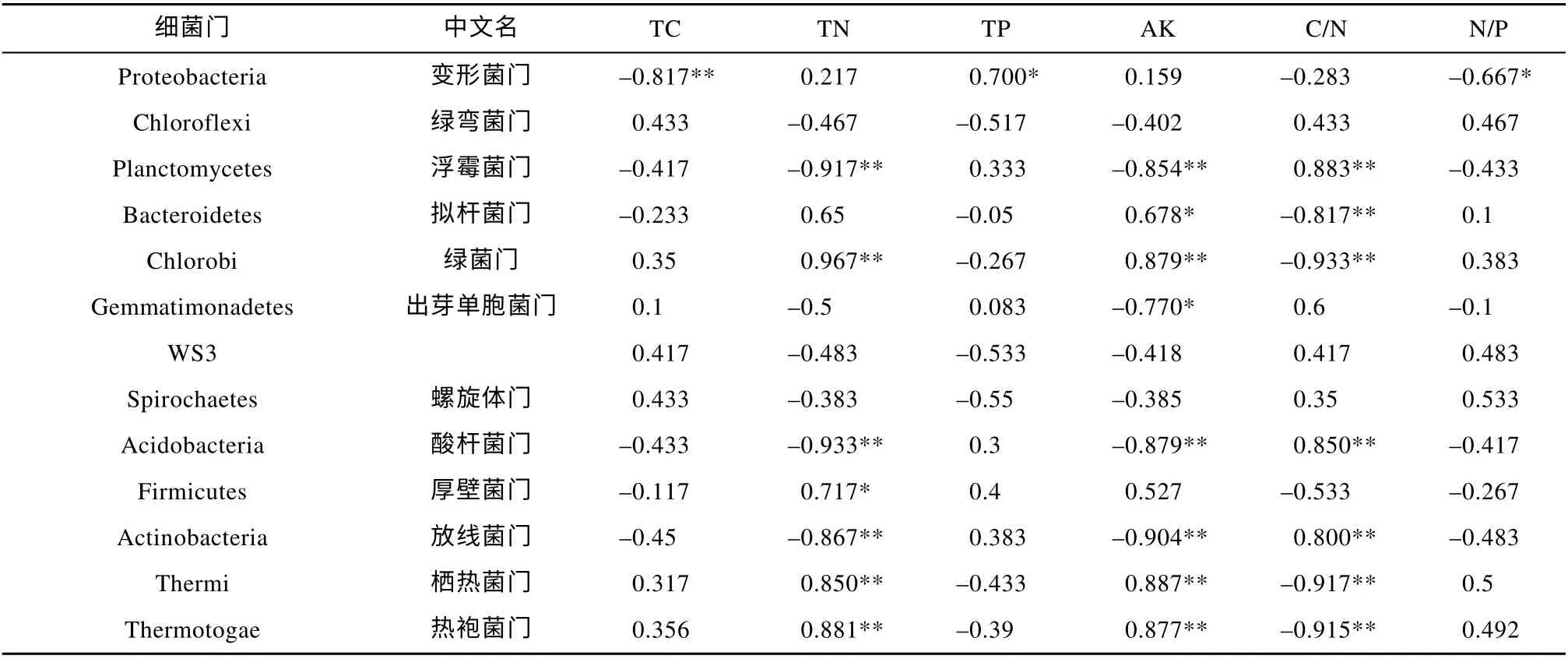
表4 土壤理化性质与门水平主要细菌的相关性Tab. 4 Correlation between soil physical and chemical properties and main bacteria at phylum level
2.8 PICRUSt 功能预测
本研究的样本丰度≥0.1%OTU 水平下的NSTI值为0.15~0.18, 根据PICRUSt 开发者的评测结果(Langille et al, 2013)认为该预测结果较好。KEGG 功能预测分析结果显示(图5), Environmental Information Processing 中的Membrance Transport 功能基因丰度和Metabolism 中的碳水化合物、氨基酸、能量以及脂质代谢等功能基因在Sg 样品中丰度比亲本高。而COG 功能预测同样发现Metabolism 功能相关的碳水化合物、氨基酸、能量代谢等基因丰度在Sg 样品中高于亲本。RFAM 功能预测在 RFAM 数据库(http://rfam.xfam.org/)对比发现Sg 样品的RF01687(Acido-Lenti-1, 主要在变形菌门和酸杆菌门中的物种发现)、RF01383(编码一种属于谷氨酸门控离子通道家族的蛋白质, 只在变形菌门中的1 个物种发现)、RF00519(一种蛋白质编码的RNA, 全部在细菌的变形菌门的物种中发现)、RF01668(一种蛋白质编码的RNA, 全部在细菌的变形菌门的物种中发现)、RF00023(transfer-messenger, 主要在变形菌门、厚壁菌门、放线菌门的物种中发现)、RF00169(细菌小信号识别颗粒RNA, 主要在变形菌门、厚壁菌门、放线菌门、拟杆菌门的物种中发现)和 RF00010(Bacterial RNase P class A, 主要在变形菌门、厚壁菌门、放线菌门、拟杆菌门的物种中发现)等RNA 分子的丰度显著上升。
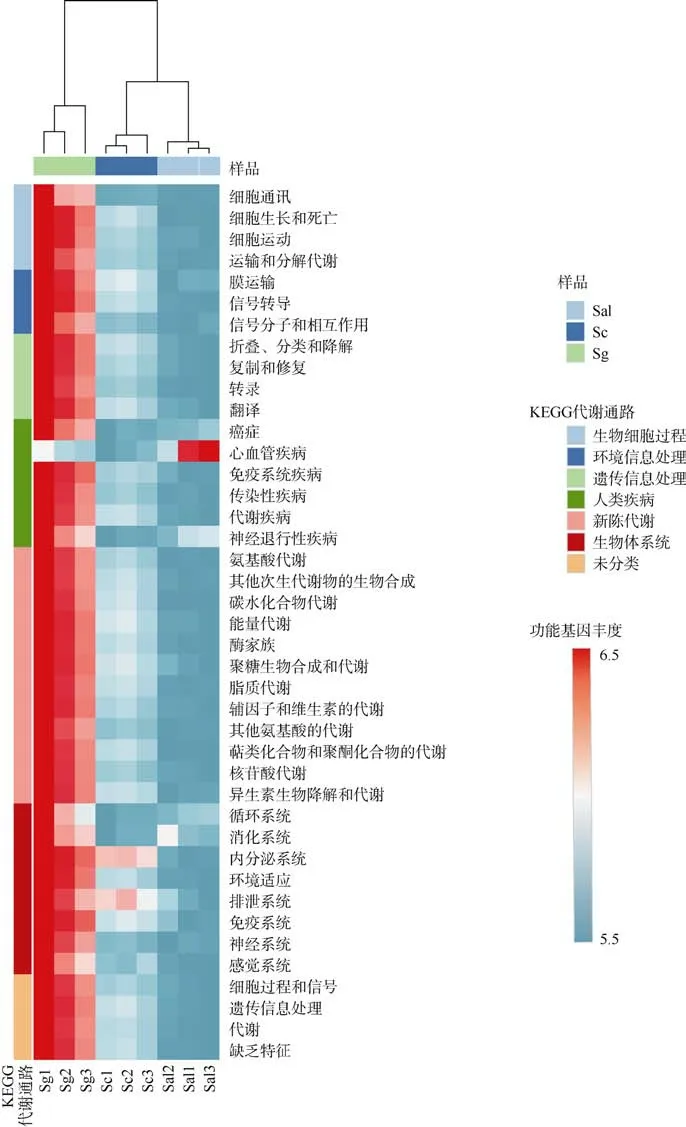
图5 KEGG 二级分类预测丰度聚类热图功能基因丰度是功能基因数目取log10 值所得Fig. 5 KEGG secondary classification prediction of abundance clustering heat map Functional gene abundance is obtained by taking the log10 value of the number of functional genes
3 讨论
红树林已经适应了潮间带—陆地和海洋生态系统之间的界面。研究表明, 红树林在生理、生态和基因组水平上具有适应性进化(Feng et al, 2020)。以往学者们对于红树植物的研究大多集中于分类、分布、生理生态、用途及价值等方面(李海生, 2003), 对海桑属红树植物的研究多关注无瓣海桑的引种及其所造成的影响(Ren et al, 2009, 2010; 曹雷雷 等,2015, Yu et al, 2020)。在微生物水平上研究区域的红树林湿地的沉积物的微生物群落结构、污染物对微生物群落的影响、参与生物化学循环的功能菌等
(Bharathi et al, 1991; Brito et al, 2006; Tian et al,2008; Zhou et al, 2009; Jiang et al, 2013)。本文采用高通量测序技术, 对具有亲缘关系的三种海桑属红树植物: 海桑(Sc)、杯萼海桑(Sal)、拟海桑(Sg)的根际土壤细菌种类进行鉴定, 分析细菌群落结构、菌群多样性, 预测细菌的生物学功能。认识了海桑属红树植物根际土壤细菌菌群的一般结构, 探究了具有亲缘关系的不同海桑属之间在根际细菌结构上的相似性和异质性, 并尝试从微生物角度去探讨根际微生物组成与杂交拟海桑具有的生长优势的关系。
拟海桑及其亲本三种海桑属样品的根际土壤细菌多样性较高。本研究的拟海桑及其亲本的根际细菌群落Shannon 指数为5.12~10.73, 高于任健 等(2012)对东寨港秋茄林(Shannon 指数4.15~4.96)和无瓣海桑(Shannon 指数3.3~3.6)的土壤细菌多样性研究结果, 以及陈玉军 等(2004)对海南省琼山县三江河两岸的红树植物无瓣海桑(Shannon 指数为1.18~1.88)、海桑(Shannon 指数为1.29~2.22)、秋茄(Shannon 指数为0.70~1.33)三种人工林群落的系统研究结果。
三种海桑样品的根际细菌群落结构有较高的相似性。变形菌门细菌广泛生活在土壤、海水和滩涂淤泥中(王勇忠, 2015; 吴冬梅, 2018, 柳旭, 2019;张起畅 等, 2020)。变形菌门在各海桑样本的根际细菌群落相对丰度均超过40%, 为绝对优势门。与任健等(2012)对无瓣海桑林下土壤细菌组成的研究结果相似, 但是低于张起畅 等(2020)研究海南东寨港红树林淤泥中的变形菌门占比为83.78%的水平。各海桑样本除优势门变形菌门外, 其余丰度丰富菌群在组成和丰度上均具有明显差异。其他较丰富的类群包括: 浮霉菌门、绿弯菌门、拟杆菌门、酸杆菌门、厚壁菌门等。除变形菌门外, 其余的丰度丰富菌群的丰度均在拟海桑和海桑样品间存在显著差异。本研究发现亲本杯萼海桑(Sal)的丰度丰富菌群种类少但是占比高, 具有高优势, 这是造成Sal 较Sc、Sg菌群多样性偏低的主要原因。杯萼海桑的栖热菌门的OTU 属于楚帕氏菌科(Trueperaceae), g_B-42 属,g_B-42 属的具体功能尚未清楚, 楚帕氏菌科细菌被证实为堆肥过程中的降温期和成熟期的标志微生物,具有碳代谢和氨基酸代谢的重要功能(Zhang et al,2020)。拟海桑及其亲本样品的根际土壤细菌在菌群多样性和结构方面对比三亚湾海草根际微生物(周卫国 等, 2019)和三沙湾沙滩微生物(王勇忠, 2015)发现, 优势门变形菌门在海草根际微生物和沙滩中占比最高, 海草为70.87%, 沙滩为50.0%, 海草的Shannon 指数为7.74, 故拟海桑和海桑样品的根际土壤细菌在菌群多样性比海草高。而杯萼海桑则比海草低。子代拟海桑的根际细菌菌群结构与亲本具有差异, 表明尽管根际微生物能从亲本传递给子代,但会发生过滤, 在过滤作用下, 亲本的个别丰度丰富菌群在子代的丰度显著降低, 甚至消失。
拟海桑及其亲本的根际细菌的群落物种组成的异质性不高。利用物种丰度划分的方法寻找可能造成异质性的关键种, 发现条件稀有或丰富类群(CRAT)的OTUs 来自丰度占优势的变形菌门、厚壁菌门、拟杆菌门、热袍菌门和栖热菌门。三元图对CRAT 的OTUs 进行贡献度分析发现对拟海桑及其亲本根际细菌群落区分贡献度大的OTU 来自变形菌门、厚壁菌门和拟杆菌门。说明在细菌群落物种组成上, 三种样品间存在着能区分群落的OTU 但是数量较少。
子代拟海桑根际微生物群落中放线菌门(Actinobacteria)和酸杆菌门(Acidobacteria)的丰度显著升高。放线菌门是土壤中分布最广的细菌门之一,以其在体外降解植物残体的能力而著称, 能促使土壤中的动物和植物遗骸腐烂(Bao et al, 2021)。Li 等(2018)研究发现放线菌门的根际促生菌可以产生吲哚乙酸(indole-3-aceticacid, IAA), 丰富的根际微生物之一节杆菌(属于放线菌门)已被报道具有产生IAA 促进植物生长的能力。酸杆菌门(Acidobacteria)是一类重要的功能菌。多项研究发现酸杆菌门(Acidobacteria)在重要生态过程中的动态作用, 即调节生物地球化学循环、分解生物聚合物、分泌胞外多糖和促进植物生长, 还具有多种代谢途径所涉及的基因, 这些基因可能有助于根际存活和竞争定殖(王光华 等, 2016; Kalam et al, 2020)。本研究通过PICRUSt 功能预测发现Sg 样本的碳水化合物、氨基酸、能量以及脂质代谢等与代谢相关的功能基因丰度明显高于亲本。酸杆菌门和放线菌门细菌的丰度增加, 使得相关功能基因增加, 并导致拟海桑的根际细菌代谢能力增强, 代谢产物增多, 供养增多,能为拟海桑生长提供更多的营养。这提高了拟海桑的生存能力, 使其具有生长优势。
土壤理化性质的测定中, 海桑和杯萼海桑的土壤全氮量比拟海桑的数值高 3 倍, 对比任健 等(2012)同一地区的无瓣海桑林的土壤理化性质, 发现本研究的根际土壤样品的TN、TP 等含量均增加。Lu 等(2021)模拟氮沉降对黄河三角洲滨海湿地土壤微生物群落多样性的影响研究发现, TN 和TC 的含量是影响土壤细菌多样性的最重要因素, 模拟TN含量增高会导致土壤细菌多样性下降, 人为提高TN 含量会导致富营养类群(变形菌门)丰度提高, 降低贫营养类群(酸杆菌)的丰度。此外多项研究发现高的TN 含量会抑制或降低微生物菌群的多样性(Van Diepen et al, 2010; 袁颖红 等, 2013; Li et al,2016; 沈芳芳 等, 2019; 林婉奇 等, 2020)。本研究的客观结果是Sg 样本的TN 含量显著低于亲本, 且其变形菌门的丰度与亲本比较并无显著降低, 相反其酸杆菌门的丰度还明显增高。所以拟海桑TN 含量降低不是人为因素导致的。Wang 等(2010)从红树植物中分离出使实验植物对N、P 和K 的吸收增加的AMF, 故拟海桑放线菌门和酸杆菌门细菌丰度的增加也可能促进拟海桑对N 元素的吸收增加。研究还发现植物分泌的化感物质(如酚类、酸类物质)也有助于植物吸收 N、P 以及金属离子等营养成分(Chapin III, 1995; Bradley et al, 1997)。齐璐(2019)研究表明Proteobacteria 在有机基质降解方面作用巨大,并在其中已发现多种可进行固氮的细菌, 但细菌的活性受多因素影响, 如pH 等, 而根际土壤的pH 值受植物根系的分泌物所形成的根际效应影响(张福锁 等, 1992; 朱丽霞 等, 2003)。故造成海桑与亲本间的TN 含量差异可能是植物-根际微生物互作的结果。相关性分析研究了TN 和13 项丰度丰富门细菌之间的相关关系后发现, 拟海桑样品的酸杆菌门、放线菌门丰度显著升高与TN 含量低相关。酸杆菌门和放线菌门的细菌含有的代谢功能基因丰富, 根系是植物吸收营养的场所, 根际微生物作为植物的第二基因组能够影响植物对营养物质的吸收。综上所述, 拟海桑生长优势的出现与其根际微生物的结构和功能的变化是相关联的。营养物质、根际微生物结构和代谢能力的变化等多因素综合作用可导致拟海桑的生长优势的出现。此外还有许多预计会影响微生物组成和活动的植物性状的因素, 例如根系分泌物(Zhalnina et al, 2018)和根系结构(Saleem et al,2018), 并且可能由大量基因控制(Deng et al, 2021)。本研究增加了我们对近缘海桑属红树植物根际细菌群落结构组成及其生态功能的了解。
在分析亲缘关系对微生物群分类组成的影响时, 由于16S rRNA 基因测序相对简单且经济实惠,因此本研究使用这一当前广泛应用的标准方法进行量化。然而, 由于rRNA 基因变异的分辨率不足和分类群之间的功能冗余, 该技术无法有效区分对宿主有功能影响和没有功能影响的微生物, 并面临着一系列问题和挑战(黄志强 等, 2021)。未来在相关研究中, 期待能发展、完善或发现更有效的方法。
猜你喜欢
中国土壤与肥料(2021年5期)2021-12-02 01:06:26
海洋通报(2020年5期)2021-01-14 09:27:04
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
水生生物学报(2019年4期)2019-07-20 08:08:10
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
天然产物研究与开发(2018年3期)2018-05-07 06:38:35
中国蔬菜(2016年8期)2017-01-15 14:23:38
发明与创新(2015年6期)2015-12-26 11:17:18
发明与创新·中学生(2015年6期)2015-06-01 00:40:24
