
金合欢素纳米混悬剂的制备及其体内药动学研究
2022-12-03 11:58张佩琛王涛
中成药 2022年11期
张佩琛王涛
(1.郑州澍青医学高等专科学校, 河南 郑州450064; 2.郑州大学, 河南 郑州450001)
金合欢素是一种黄酮类化合物,广泛存在于洋槐树、金合欢树、密蒙花、菊花、大蓟等植物的根茎中,具有抗氧化、抗肿瘤、抗菌、心脏保护、抗骨质疏松、抗动脉粥样硬化、抗炎、免疫调节等作用[1⁃4],但该成分溶解度仅为36.43 μg/mL[5],会影响其溶出速率及溶出度,而且口服生物利用度仅为2.34%[6],又会限制药效发挥。胡瑞瑞等[7]制备了金合欢素聚乳酸纳米粒,但其载药量仅为6.08%。
纳米混悬剂无需载体材料即有较高的载药量,并且药物溶解度、溶出度、生物利用度、药效等参数均可明显改善[8⁃10],在医药领域应用潜力较大,工业化程度较高。因此,本实验将金合欢素制成纳米混悬剂,采用Box⁃Behnken 响应面法优化该工艺,并考察其体内药动学,以期为该制剂后续开发利用提供参考。
1 材料
Agilent 1200 型高效液相色谱仪(美国Agilent公司);ATS 型均质机(德国 Seeker公司);MSE125P⁃CE 型电子天平(配置防风罩,德国Sartorius 公司);HJ⁃2 磁力加热搅拌器(青岛创聚环保集团有限公司);Nano⁃S90 型粒度分析仪(英国马尔文仪器有限公司);FD⁃1C⁃50 型实验室冷冻干燥机(上海贺帆仪器有限公司);QIMO⁃DCY⁃12 S 型氮气吹扫仪(上海琪摩电子科技有限公司)。
金合欢素对照品(批号110981⁃201910,纯度99.0%,中国食品药品检定研究院);金合欢素原料药(批号20191026,纯度97%,成都嘉叶生物科技有限公司)。十二烷基磺酸钠(SDS,批号171114,国药集团化学试剂有限公司);氯磺丙脲(批号200218,美国Sigma 公司);PVP K30(批号190506)、泊洛沙姆188(批号181225)(安徽山河药用辅料有限公司)。
SD 大鼠,体质量(240±20)g,购自河南省动物实验中心,动物生产许可证号SCXK(豫)2016⁃0001,饲养于温度、相对湿度分别为(25±2)℃、(45±5)%的环境下。
2 方法与结果
2.1 纳米混悬剂制备 取50 mg 金合欢素原料药,加入30 mL 有机溶剂(四氢呋喃∶乙醇=1∶1),65 ℃、1 000 r/min 磁力搅拌至溶解,作为有机相;称取一定量PVP K30、SDS,加入100 mL 蒸馏水,65 ℃、1 000 r/min 磁力搅拌至溶解,作为水相,将有机相缓慢滴加到水相中,在45 ℃下旋转蒸发30 min 除去有机溶剂,在均质压力1 000 bar(1 bar=100 kPa)下循环均质数次,加入蒸馏水至100 mL,过0.45 μm 微孔滤膜,即得。
2.2 粒径测定 取0.1 mL 纳米混悬剂,加入4 mL蒸馏水混匀,取适量至比色皿中,置于粒度分析仪中测定粒径。
2.3 Box⁃Behnken 响应面法 固定金合欢素用量为50 mg,稳定剂为PVP K30、SDS 混合物,选择PVP K30 占比(X1)、稳定剂用量(X2)、均质次数(X3)作为影响因素,粒径(Y)作为评价指标,Box⁃Behnken 响应面法优化制备工艺,因素水平见表1,结果见表2。
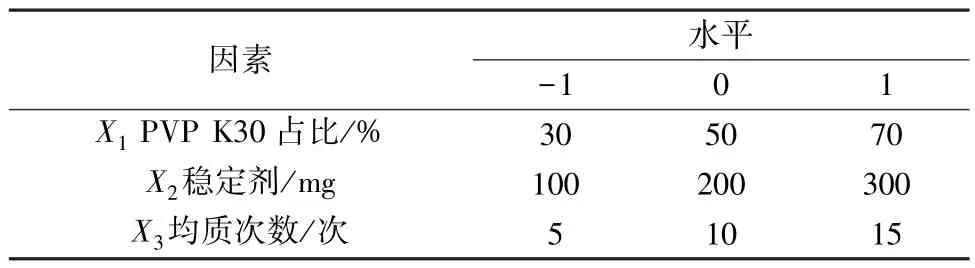
表1 因素水平Tab.1 Factors and levels
对表2 数据进行拟合,得二次多元回归方程为Y=214.62+10.100X1-25.94X2-35.84X3+19.38X1X2-46.93X1X3-5.55X2X3+19.61+71.39+77.89,R2=0.997 5,AdjR2=0.994 2,表明模型拟合度良好,方差分析见表3。由此可知,模型P<0.01,具有高度显著性;失拟项P>0.05,表明未知因素干扰很小,模型可信度较高,可用于优化;因素X2、X3、X1X2、X1X3、有极显著影响(P<0.01)。

表2 试验设计与结果Tab.2 Design and results of tests

表3 方差分析Tab.3 Analysis of variance
采用Design Expert V8.0.6 软件进行响应面分析,结果见图1。由此可知,均质次数不变时,随着PVP K30 占比或稳定剂用量增加,粒径先减小后增大,可能是由于两者不足时会影响稳定效果,导致纳米粒之间发生融合、聚集等现象,使得粒径较大,但两者过量时稳定剂会吸附在纳米颗粒表面,粒径也会变大[10];稳定剂用量不变时,随着均质次数增加,粒径先减小后增大,可能是由于均质次数过少时不足以形成较小粒径的纳米混悬剂,但过多时会破坏纳米粒稳定剂保护层[8],进而发生融合、聚集等,导致粒径变大;PVP K30 占比不变时,随着均质次数增加,粒径先减小后增大,可能是由于均质次数过多时纳米混悬剂温度较高[8],影响了体系稳定性,导致粒径变大。最终确定,最优工艺为PVP K30 占比47.16%,稳定剂用量215.04 mg,均质次数11.92次,粒径为209.8 nm,为便于操作,将其修正为PVP K30 占比47%,稳定剂用量215 mg,均质次数12 次。

图1 各因素响应面图Fig.1 Response surface plots for various factors
2.4 HPLC 法测定金合欢素含量
2.4.1 色谱条件 Syncronis C18色谱柱(250 mm×4.6 mm,5 μm);流动相乙腈⁃0.2% 磷酸(45∶55);体积流量1.0 mL/min;柱温35 ℃;检测波长268 nm;进样量20 μL。
2.4.2 线性关系考察 称取金合欢素对照品10 mg至100 mL 量瓶中,甲醇超声溶解并定容,得100 μg/mL 贮备液,流动相依次稀释成20、10、1、0.5、0.1、0.05 μg/mL 对照品溶液,在“2.4.1”项色谱条件下进样测定。以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y)进行回归,得方程为Y=13.645 9X+1.426 7(r=0.999 9),在0.05~20 μg/mL范围内线性关系良好。
2.4.3 供试品溶液制备 取1 mL 纳米混悬剂,置于50 mL 量瓶中,加入30 mL 甲醇超声处理5 min,甲醇定容至刻度,过0.45 μm 微孔滤膜,取续滤液,即得。
2.4.4 方法学考察 取同一份纳米混悬剂,按“2.4.3”项下方法平行制备6 份供试品溶液,在“2.4.1”项色谱条件下进样测定,测得金合欢素峰面积RSD 为0.67%,表明该方法重复性良好。取同一份供试品溶液,于0、3、6、9、12、24 h在“2.4.1”项色谱条件下进样测定,测得金合欢素峰面积RSD 为0.58%,表明溶液在24 h 内稳定性良好。取20、1、0.05 μg/mL 对照品溶液适量,在“2.4.1”项色谱条件下各进样测定6次,测得金合欢素峰面积RSD 分别为0.36%、0.29%、0.44%,表明仪器精密度良好。取9 份供试品溶液,每份0.5 mL,平均分为3组,每组3份,分别加入“2.4.2”项下贮备液1.5、2.5、3.5 mL,在“2.4.1”项色谱条件下进样测定,测得金合欢素平均加样回收率分别为101.16%、99.27%、100.54%,RSD 分别为1.68%、0.61%、0.83%。
2.5 验证试验及载药量测定 按“2.3”项下优化工艺平行制备3 批纳米混悬剂,分别取0.1 mL,加入4 mL 蒸馏水混匀,测定粒径、Zeta 电位。结果,平均粒径为214.7 nm(图2),PDI 为0.094,Zeta 电位为-31.6 mV(图3)。

图2 金合欢素纳米混悬剂粒径分布Fig.2 Particle size distribution of acacetin nanosuspensions

图3 金合欢素纳米混悬剂Zeta 电位Fig.3 Zeta potential of acacetin nanosuspensions
取1 mL 纳米混悬剂至50 mL 量瓶中,加入30 mL甲醇超声处理5 min,甲醇定容至刻度,过0.45 μm 微孔滤膜,取续滤液,在“2.4.1”项色谱条件下进样测定,计算金合欢素含量(M2),平行3 次;另取1 mL,在-40 ℃下预冻48 h 后真空低温冻干得粉末,称定质量(M1),平行3次,计算载药量,公式为载药量=(M2/M1)×100%。结果,平均载药量为18.77%。
2.6 冻干粉制备及表征
2.6.1 制备工艺 取纳米混悬剂适量,加入6%甘露醇振荡溶解,取3 mL 至西林瓶中,在-40 ℃下预冻48 h,迅速置于-20 ℃冷冻干燥机中抽真空,冻干48 h 后取出,即得,立即密封并置于干燥器中保存,外观见图4。取3 批冻干粉,蒸馏水复溶,测得其平均粒径为248.14 nm。

图4 冻干粉外观Fig.4 Appearance of lyophilized powder
2.6.2 溶解度测定 取过量原料药、物理混合物(PVP K30+SDS,比例同最优工艺)、纳米混悬剂冻干粉适量置于烧杯中,加蒸馏水,室温1 000 r/min磁力搅拌3 d,取上层混悬液,8 500 r/min 离心20 min,取上清液,在“2.4.1”项色谱条件下进样测定,计算溶解度,结果见图5。由此可知,物理混合物将原料药溶解度提高至1.22倍,而纳米混悬剂可提高至11.71 倍。

图5 各样品溶解度(n=3)Fig.5 Solubilities of various samples(n=3)
2.6.3 沉降体积比考察 取原料药、物理混合物、纳米混悬剂适量,置于50 mL 具塞量筒中,振荡1 min,记录初始高速(H0),再于室温环境(温度25 ℃、相对湿度55%)下静置3 h,记录最终高度(H),计算沉降体积比,公式为沉降体积比=H/H0。结果,纳米混悬剂未发生沉淀,沉降体积比为1,而原料药、物理混合物均下降至0.62。
2.7 体外溶出研究 取原料药、物理混合物、纳米混悬剂冻干粉适量(以金合欢素计均为10 mg),加入4 mL 蒸馏水制成混悬液,置于活化后的透析袋中(截留分子量8 000~14 000 Da),细尼龙绳将两端扎紧,选择900 mL 0.5%SDS 溶液作为溶出介质,在温度37 ℃、转速100 r/min 条件下于0.25、0.5、1、1.5、2、2.5、3、4、5、6、9、12 h各取样3 mL,同时补加3 mL 空白溶出介质,过0.45 μm 水相微孔滤膜,在“2.4.1”项色谱条件下进样测定,计算溶出度,结果见图6。由此可知,原料药12 h 内累积溶出度不足40%;物理混合物12 h 内累积溶出度也仅提高至47.07%;纳米混悬剂6 h 内累积释放度接近90%,12 h 内达95.82%。

图6 金合欢素体外溶出曲线(n=3)Fig.6 In vitro dissolution curves for acacetin(n=3)
2.8 体内药动学研究
2.8.1 HPLC⁃MS 分析条件 ZORBAX SB C18色谱柱(2.1 mm×100 mm,1.8 μm);流动相乙腈⁃0.1%甲酸(20∶80);体积流量0.2 mL/min;柱温30 ℃;进样量5 μL;电喷雾离子源,负离子扫描;毛细管电压3 000 V;雾化气压力30 psig(1 psig =68.194 8 kPa);干燥气体积流量8 L/min,温度325 ℃;定量分析离子m/z285.22(金合欢素)、m/z277.60(氯磺丙脲,内标)。
2.8.2 分组、给药及血浆处理 取原料药、物理混合物、纳米混悬剂冻干粉末适量,制成0.5%CMC⁃Na 混悬液,作为灌胃液(以金合欢素计,质量浓度为7 mg/mL)。取18 只禁食12 h 的大鼠,随机分为3组,每组6只,按50 mg/kg 剂量灌胃给药。乙醚麻醉大鼠后,原料药组于0、0.167、0.25、0.5、1、1.5、2、3、4 h 采血,物理混合物组增加5 h 取血点,纳米混悬剂组增加5、6、8 h取血点,分别取约0.2 mL,置于肝素化离心管中,3 000 r/min 离心3 min,得含药血浆,吸取50 μL,加入10 μL 内标溶液(将含100 μg/mL 乙腈的氯磺丙脲对照品母液用乙腈稀释至400 ng/mL,即得)、1.5 mL 乙腈,密封涡旋6 min,8 000 r/min离心5 min,取上层有机相,45 ℃氮气缓慢吹干,加入50 μL 甲醇超声处理3 min,进样分析。
2.8.3 线性关系考察 取500 ng/mL 对照品溶液,乙腈逐步稀释至250、200、100、50、10 ng/mL,分别取50 μL,45 ℃氮气缓慢吹干,加入50 μL 空白血浆,密封涡旋6 min,即得血浆对照品溶液,按“2.8.2”项下方法处理,在“2.8.1”项条件下进样测定。以对照品质量浓度为横坐标(X),金合欢素、内标峰面积比值为纵坐标(Y)进行回归,得方程为Y=0.057 1X+0.305 4(r=0.994 2),在10~500 ng/mL 范围内线性关系良好。
2.8.4 专属性考察 取空白血浆、内标+灌胃给药4 h 后血浆、内标+血浆对照品溶液(10 ng/mL)适量,在“2.8.1”项条件下进样测定,结果见图7。由此可知,金合欢素色谱峰与其他杂质峰分离度理想,表明该方法专属性良好。

图7 金合欢素提取离子流色谱图Fig.7 Extraction ion current chromatograms of acacetin
2.8.5 方法学考察 取10、200、500 ng/mL 质控样品溶液适量,按“2.8.2”项下方法处理,在“2.8.1”项条件下进样测定,计算峰面积(ρ1);取50 μL 空白血浆,按“2.8.2”项下方法处理(不加内标)得残渣,加入10、200、500 ng/mL对照品溶液及内标溶液,在“2.8.1”项条件下进样 测定,计算峰面积(ρ2);取 10、200、500 ng/mL质控样品溶液(不按“2.8.2”项下方法处理),在“2.8.1”项条件下进样测定,计算峰面积(ρ3),根据公式(ρ2/ρ3)×100% 计算基质效应,(ρ1/ρ2)×100%计算提取回收率,结果低、中、高质量浓度金合欢素基质效应分别为96.15%、93.27%、94.55%,提取回收率分别为90.69%、94.74%、89.07%,而内标两者分别为95.17%、93.21%,表明该方法基质效应较小。取灌胃给药1 h后血浆,于0、2、4、6、8、12、24 h在“2.8.1”项条件下进样测定,测得金合欢素、内标峰面积比值RSD 为11.01%,表明血浆在24 h 内稳定性良好。取10、200、500 ng/mL 血浆对照品溶液,同一天内在“2.8.1”项条件下各进样测定6次,测得金合欢素、内标峰面积比值RSD分别为9.61%、6.28%、8.17%,表明日内精密度良好,再根据随行标准曲线计算金合欢素含量,测得其准确度分别为107.52%、95.38%、103.91%;同法每天各测定6次,连续6 d,测得两者比值RSD 分别为11.73%、8.64%、9.84%,表明日间精密度良好,准确度分别为103.66%、108.92%、97.16%。
2.8.6 结果分析 采用实测值计算tmax、Cmax,梯形法计算AUC,结果见表4,血药浓度⁃时间曲线见图8。由此可知,与原料药比较,物理混合物Cmax、AUC0~t、AUC0~∞升高(P<0.05),表明辅料增溶作用促进了药物吸收,但作用有限,相对生物利用度仅提高至1.33 倍;纳米混悬剂tmax缩短(P<0.01),Cmax、AUC0~t、AUC0~∞升 高(P<0.01),相对生物利用度提高至5.18倍,程度远大于物理混合物。
表4 金合欢素主要药动学参数(,n=6)Tab.4 Main pharmacokinetic parameters for acacetin(,n=6)

表4 金合欢素主要药动学参数(,n=6)Tab.4 Main pharmacokinetic parameters for acacetin(,n=6)
注:与金合欢素比较,∗P<0.05,∗∗P<0.01。

图8 金合欢素血药浓度⁃时间曲线(n=6)Fig.8 Plasma concentration⁃time curves for acacetin(n=6)
3 讨论
PVP K30 与SDS 联合应用作为稳定剂时,纳米混悬剂粒径较小,可能是由于前者黏度较大,可有效抑制纳米粒子的碰撞,并且分子链较长,提供了较大的空间位阻效应[11⁃13];后者是一种阴离子表面活性剂,可有效降低界面张力,有利于在制备过程中降低粒径[11],但稳定剂总用量、PVP K30 占比对粒径影响较大,故结合预实验对两者(100~300 mg、30%~70%)进行优化。另外,均质压力过小或过大均会导致粒径变大,故本实验在均质压力100 000 kPa 的前提下对均质次数(5~15 次)进行优化。
据文献[2⁃3] 报道,大鼠口服金合欢素剂量为10~80 mg/kg时即有明显的神经保护、降血脂、抗动脉粥样硬化等作用,故本实验选择50 mg/kg进行药动学研究。结果,金合欢素纳米混悬剂大大降低了药物粒径,药物溶出速率明显提高[13⁃14],导致tmax显著缩短,Cmax升高,可能与该剂型可有效改善药物溶解度、累积溶出度有关[15⁃16]。研究表明,纳米混悬剂可增加药物比表面积,有利于其充分吸收[17⁃20];纳米药物具有较强的胃肠道渗透性[17],可增加经胞间、淋巴循环等途径进入血液循环[21]。本实验发现,将金合欢素制成纳米混悬剂后生物利用度提高至5.18 倍。今后,将对金合欢素纳米混悬剂的注射药动学、毒性、药效学等指标作进一步研究。
猜你喜欢
江西化工(2022年3期)2022-07-21
上海理工大学学报(2021年3期)2021-07-20
天津中医药(2020年6期)2020-06-23
中成药(2019年10期)2019-10-24
中国盐业(2018年20期)2019-01-14
西南石油大学学报(自然科学版)(2018年1期)2018-02-10
铁道科学与工程学报(2015年5期)2015-12-24
医学研究杂志(2015年12期)2015-06-10
中国当代医药(2015年33期)2015-03-01
中华皮肤科杂志(2014年4期)2014-12-19
