
烟煤物理吸附氧气的吸附机理
2022-12-01 08:53:14华坤鹏逯园坤
煤矿安全 2022年11期
华坤鹏,逯园坤
(1.中煤科工集团常州研究院有限公司,江苏 常州 213015;2.天地(常州)自动化股份有限公司,江苏 常州 213015;3.辽宁工程技术大学 安全科学与工程学院,辽宁 葫芦岛 125105)
煤炭是我国的主要能源,占能源结构的70%。煤炭自燃是威胁煤矿安全生产的重大灾害之一。虽然从17 世纪以来己经有大量关于自燃的研究,但是目前还没有清楚地了解煤的自燃机理。煤自燃机理的研究工作,对于提出行之有效的防治措施,提高自燃防治工作的效率非常重要,对保护矿工人身安全、保护煤矿资源和净化生态的环境具有重要的实用价值[1-3]。
近年来,为了从分子水平上认识煤与氧气的物理吸附的相互作用机制,许多学者分别从物理实验和量子化学方法进行了研究。在通过煤与氧气物理吸附的物理实验来研究其特性方面,邓军等[4]利用ZRJ-1 型煤自燃倾向测定仪研究了不同温度下的煤的物理吸氧量变化规律;张树川等[5]采用ZRJ-1 型煤自燃倾向测定仪在不同氧气流量下煤物理吸附氧进行了实验研究,得到在环境温度等条件相同的情况下煤物理吸内氧量的最佳氧气流量,从而确定了煤自燃倾向性鉴定实验的最适合吸附氧气流量;一系列结果表明,影响吸氧量的因素有温度、粒度、水分等。另一方面,通过分子模拟方法也是对煤与氧气的物理吸附的一种比较好的方式,邓存宝等[6-7]提出了煤表面在侧链吸附单氧分子,要比在苯环上吸附对煤的氧化自燃的贡献大、氧分子更易与煤表面中的氨基发生物理吸附;吴康华[8]整理了甲基苯甲酸模型、甲基苯甲醛、苯甲醚、苯甲醇的键长、Mulliken 电荷布居数、吸附能得到了氧分子在活性基团上发生吸附时,将氧分子在不同活性基团上的吸附活性排序:甲氧基>醛基>带经基的亚甲基>甲基>竣基;王晓波等[9]以简化的芳香环作为煤的芳香环骨架,并适当添加侧链构建煤活性特征结构简化模型,用于分析煤吸附氧气的作用机理,通过比较几何构型、吸附位置、吸附平衡距离、电荷集居数与振动频率、吸附能得到侧链对氧分子的吸附会造成苯环吸附减弱;相建华等[10]应用巨正则系综蒙特卡洛(GCMC)及分子动力学方法对兖州煤模型的吸附行为进行了研究,获得了CH4、CO2与H2O 的吸附量、吸附构型以及含氧官能团的影响,并利用等量吸附热及能量变化数据揭示了3 种物质的不同吸附机理;金智新等[11]采用蒙特卡洛方法得到了水在煤中吸附过程概率密度的三维分布,从而更直观全方位地认识水的吸附行为及机理;武司苑等[12]应用用Materials Studio 软件得出一些CO2、N2等气体的吸附规律,但Gaussian 程序是专门做量子力学的软件,对分子动力学的模拟缺少一些动力学的分析。
从以上的研究中,可以发现目前对煤物理吸附和化学吸附的研究现状,还缺少煤物理吸附O2量的范围和影响因素研究;同时对氧气的吸附研究不是特别的完善;且在实验中煤对氧气物理吸附和化学吸附的界限无法区分,无法通过实验方式把化学吸附和物理吸附氧气彻底区分开。为了进一步探究煤自燃机理,更好地掌握自燃的规律,应当加大对煤物理吸附氧气的模拟研究力度,同时也应对煤物理吸附氧气微观变化加大研究力度。为此,利用Materials Studio 软件的蒙特卡洛模拟来进行煤氧物理吸附研究,包括吸附的范德华能、等量吸附热、吸附量、吸附密度、径向分布函数、均方位移等;从微观角度研究煤分子对氧分子的物理吸附过程,分析影响煤氧吸附的相关因素,得到煤物理吸附氧气的规律,以了解煤物理吸附O2的特性[13-16]。
1 煤结构模型与计算方法
1.1 平顶山烟煤模型
煤结构十分复杂,没有固定的模型。以平顶山烟煤为对照,建立了煤分子模型,分子式为C108H71O4N,C、H、O、N 的质量分数分别为89.69%、4.91%、4.43%、0.97%。与平顶山烟煤的元素分析的质量分数十分接近。
首先建立煤基本结构和甲烷分子,利用采用Amorphous Cell 模块根据基本结构单元固定密度构建周期性结构,将9 个煤分子和900 个甲烷分子共同构筑在晶胞内,构筑密度为0.3 g/cm3,力场选择Dreiding 力场,温度为298 K,计算步数为1×107步,然后将900 个甲烷分子删除。通过Forcite 模块进行几何优化,首先进行结构优化,静电场利用Atom based,设置范德华力的截断半径小于煤晶胞的一半,力场选择Dreiding 力场,计算步数为1×106,计算得出初步的煤超晶胞,首先需要宏观正则系综(NPT)进行人为扩胞,力场选择Dreiding 力场,模拟步数设为5×106,帧数间隔步数为500,温度为298 K,使内应力变的合理,由于扩胞后煤分子会发生一些扭曲,故需要用等温等压系统(NVT)使煤分子充分舒展开,力场选择Dreiding 力场,模拟步数设为5×106,帧数间隔步数为500,温度为298 K,由于NPT 使体系具有一定的速度,所以初始速度设为Current,但得出的煤晶胞有可能没收敛,需要重复进行NPT 和NVT,使煤晶胞的边长趋于稳定不变,在经过2 次循环可得出合适的超晶胞。煤结构及O2结构模型如图1。
对于O2的模型现阶段有2 种模型:一种是非极性分子,原子电荷为0;另1 种是带虚原子的O2模型,如B Vujic[17]建立的O2模型(图1),通过O-mid来构造O2模型,这种模型的主要目的是两端的氧原子点带电荷,但整体的氧分子的是不带电荷。选择更合理的非极性分子O2模型即氧分子的原子电荷为0,O-O 键长为0.148 0 nm。
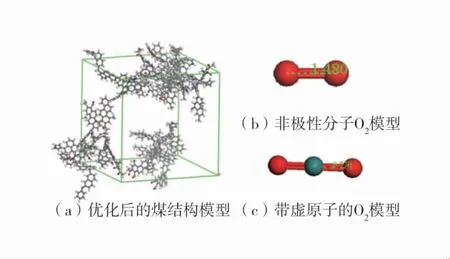
图1 煤结构及O2 结构模型Fig.1 Coal structure and oxygen structure model
1.2 计算方法
利用Materials Studio 中的Sorption 模块,采用巨正则系综蒙特卡洛(grand canonical monte carlo GCMC)方法,把吸附质分子看作刚性小分子,因此,采用Metropolis 接受规则,接受吸附质分子交换、构象异构化、转动、平动和再生的比例分别为4、2、2、2、0.1,其中转动的最大角度为5°平动最大距离为0.1 nm。模拟的力场选择Dreiding 力场,Dreiding 力场适用于有机小分子、大分子、主族元素的计算,在该力场中总能量(E)包括价电子能(Eval)和非成键能(Enb),范德华能(Evdw)和氢键作用能(Ehb)都包含在非成键能中。前人研究结果表明此力场适用于煤结构以及吸附的研究[18],静电作用采用Ewald 加和法,精度为4.186×10-3kJ/mol,范德华作用和氢键作用采用Atom based 法,范德华力的截断半径小于煤晶胞的一半即可,煤盒子的边长为38.899 nm,故分别取16 nm 和1 nm。经过计算为使结果具有依据性,在模拟中C/D 要达到1.0 左右,经过试算将模拟步数设为2×107,其中包括1×107的平衡步数,1×107的生产步数,最小能量框设为10 帧,用来计算过程中的物理吸附量。
2 结果与讨论
2.1 O2 的单组分等量吸附热
为研究温度对煤物理吸附O2的影响,分析了在298.15、303.15 K,随压强增大的等量吸附热,不同压力下等量吸附热随吸附量变化情况如图2。由图2可知:由于温度的升高,等量吸附热逐渐下降,且等量吸附热相对来说比较小,都小于42 kJ/mol,均属于物理吸附,从侧面验证了本方法适用于研究煤物理吸附氧气的特性。可以看出在模拟的情况下,温度对等量吸附热的影响相对来说变化微小,且随着压强的增大等量吸附热的变化微小。
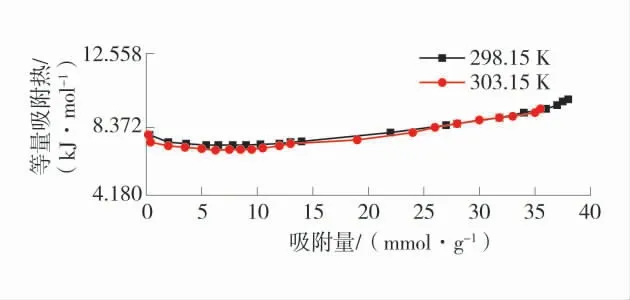
图2 不同压力下等量吸附热随吸附量变化情况Fig.2 Variation of equal adsorption heat with adsorption capacity under different pressures
2.2 烟煤吸附能量变化
除了预测宏观现象,如吸附等温线和吸附热,GCMC 模拟还可以提供分子尺度上的详细图片,这些信息可以进一步分析以获得能量相互作用、择优位置和最终吸附机理的信息。这些势能图说明了框架中具有最低相互作用能的区域,因此也就说明了优先吸附位点,故利用GCMC 模拟结果进行能量分析也是对物理吸附氧气理论的补充,吸附能量分布图如图3。图中:P(E)为能量E 的概率分布。

图3 吸附能量分布图Fig.3 Adsorption energy distribution
在压力为定值100 kPa,在283.15~363.15 K时,以能量为横轴,以E 的概率为y 轴,即P(E)是E的概率,数值越大,则该能量的概率越高,因此也就说明了优先吸附位点,负值越大说明相互作用越强,吸附位越有利。从图3 可知,温度升高,能量分布的概率越高,即在363.15 K 时P(E)在达到了最高点,即此时概率最大,此值靠近0,即相互作用稍弱,其大致规律是物理吸附O2时随温度升高,相互作用呈减弱的趋势,从而影响吸附量下降。
2.3 吸附密度场变化
为更加直观地了解煤物理吸附O2的吸附位及吸附时的微观变化,同时分析了煤物理吸附O2的密度场变化。密度场分布代表了在蒙特卡洛平衡步数中煤物理吸附氧气的吸附位的统计平均的结果,在298.15、303.15 K 下70~120 kPa 的吸附密度场变化情况如图4、图5。图中的颜色越深表示其吸附位的吸附的概率越大,可以发现在298.15 K 时,其吸附随压力增加,其着色点越多,意味着其吸附量越大,且大部分呈蓝色,分布在空腔中,表示孔隙的吸附密度很低,且随压力增加,着色点少部分由蓝转红,说明其物理吸附会随压力的增加逐渐达到饱和,也从侧面说明煤物理吸附O2不易达到饱和。
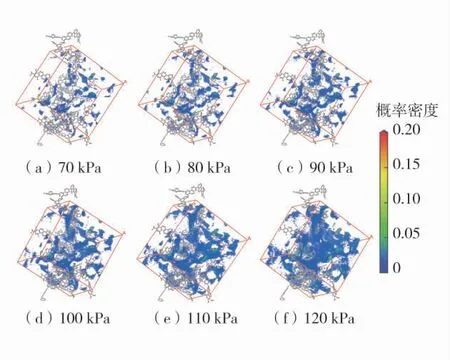
图4 298.15 K 下吸附概率密度分布图Fig.4 Distribution of adsorption probability density at 298.15 K
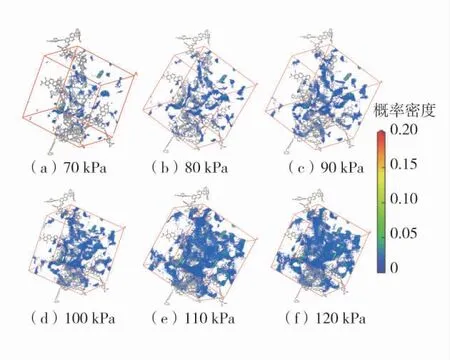
图5 303.15 K 下吸附概率密度分布图Fig.5 Distribution of adsorption probability density at 303.15 K
同时为比较温度对煤物理吸附O2的影响,对比298.15、303.15 K 的吸附密度图,发现在同等压力下,由298.15 K 至303.15 K 其着色点个数稍有降低,说明吸附位概率稍有减少,吸附量有下降。
煤物理吸附O2的密度分布的分析,与上述的等量吸附热、等温吸附线、吸附能量、吸附量得到的结果是一致,这些特性相互验证,为煤物理吸附O2机理提供更多的理论基础。
2.4 孔隙中O2 分子和官能团的相互作用
2.4.1 孔隙中O2分子和芳香官能团的相互作用
径向分布函数gab(r)(Radial distribution function)是描述粒子密度作为距参考原子的距离的函数如何变化,是表征粒子微观结构特征的物理量,能够反映粒子聚集有序的特性,可以解释为系统的局域密度与平均密度的比值,其表达式为:
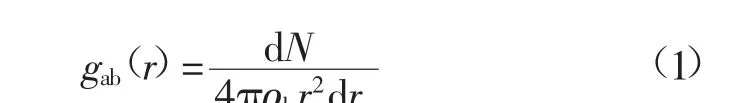
式中:dN 为距a 原子r 到r+dr 壳层范围内的b粒子的数目;ρb为粒子b 的密度。
煤中芳香结构和脂肪结构编号图如图6,在温度298.15 K 的温度下孔隙中O2与各类官能团的径向分布函数如图7。
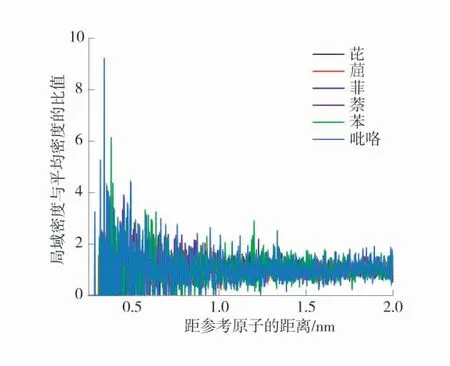
图7 298.15 K 的温度下孔隙中O2 与各类官能团的径向分布函数Fig.7 Radial distribution function of O2 and various functional groups in pores at 298.15 K
由图7 可知,O2与吡咯形成明显主峰,峰值大小为9.239 9,说明在吸附过程中,吡咯官能团的吸附能力相对几种官能团来说是最强的,其次是苯,峰值大小是6.160 4,依次可得出各个官能团的吸附能力顺序,为吡咯>苯>萘>芘>菲> ,峰值大小分别为9.239 9、6.160 4、4.029 2、2.837 0、2.581 8、2.220 6,观察其结构,发现其芳香缩聚程度是影响O2吸附的最大因素。
由于在分子结构中具有类似的芳香结构,故对比同种芳香结构但侧链不同时,对O2吸附的影响,煤中芳香结构径向分布函数图如图8。
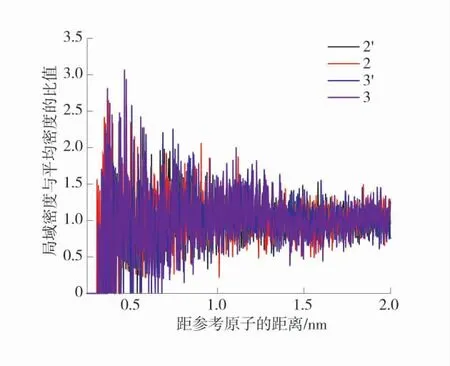
图8 煤中芳香结构径向分布函数图Fig.8 Radial distribution function diagram of aromatic structure in coal
由图8 可知,在同种结构中的峰值有不同,即3′>3,峰值分别为3.069 3、2.581 8,同时2>2′,峰值分别为2.650 0、2.315 5,对比结构发现芳香官能团对于吸附的影响强弱取决于其链长。
2.4.2 孔隙中O2分子和脂肪官能团的相互作用
根据数据分析对比并计算径向分布函数得到煤中脂肪结构径向分布函数如图9。由图9 可知,羟基侧链是对O2吸附作用较强的官能团,甲基稍弱于羟基侧链,其羟基峰值大小为13.588 1,其次为10 号甲基峰值大小为7.626 0,7、9、11 号甲基峰值大小分别为6.245 6、7.325 0、6.930 4,可知在吸附过程中羟基比甲基的作用大,说明在吸附时优先吸附羟基侧链,对比芳香结构的径向分布函数,发现芳香官能团的峰值普遍小于脂肪结构,可以分析出芳香官能团主要提供O2的栖息空间,侧链含量、空隙的开放程度、芳香官能团的大小等决定了其吸附能力,优先吸附位点为羟基。这对于抑制煤物理吸附O2作用机理细化补充提供参考。
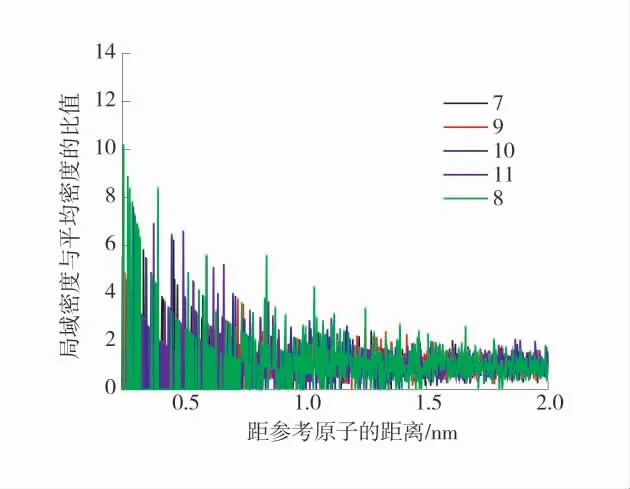
图9 煤中脂肪结构径向分布函数Fig.9 Radial distribution function of fat structure in coal
2.4.3 氧气的吸附位
利用蒙特卡洛方法与分子动力学计算得出的吸附密度场和吸附构型图分别如图10、图11。可以得出,2 种计算结果一致,在物理吸附O2过程中,O2分子大部分赋存在芳香官能团所围成的一系列微孔中。
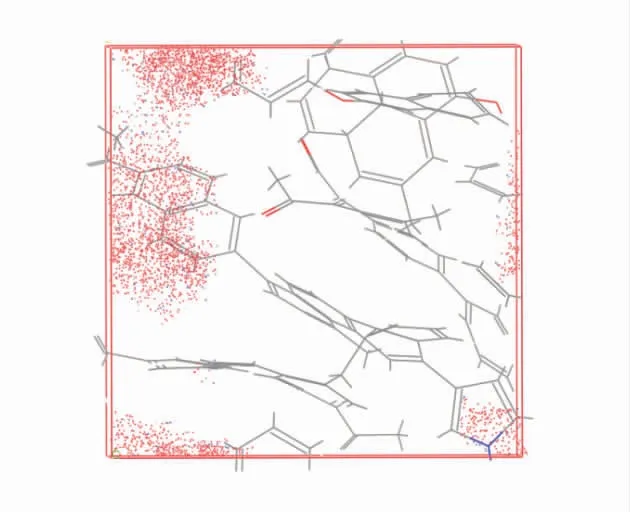
图10 蒙特卡洛方法的吸附密度场Fig.10 Adsorption density field of Monte Carlo method
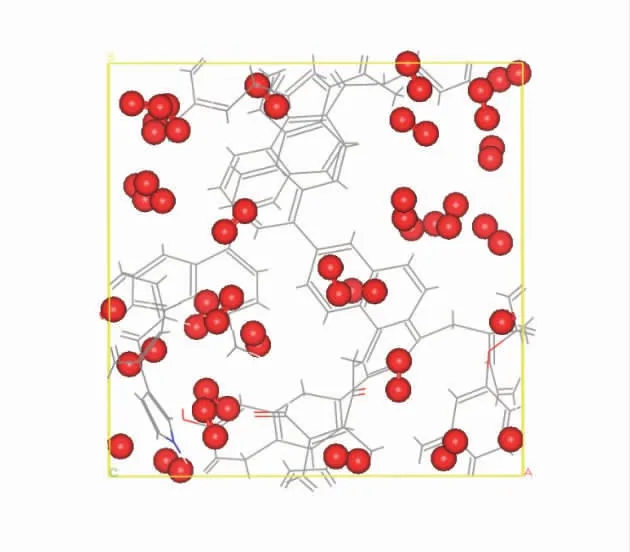
图11 分子动力学计算的吸附后的结构图Fig.11 Structure diagram after adsorption calculated by molecular dynamics
2.5 孔隙中O2 分子的扩散
通过MD 来计算粒子的微分方程得到其运动的瞬时坐标,分别统计模拟时间内的均方位移平均值,发现粒子的均方位移和时间已达到线性关系,由Enistein 定律表面扩散系数达到恒定,说明本次模拟符合扩散系数统计的可靠性要求。
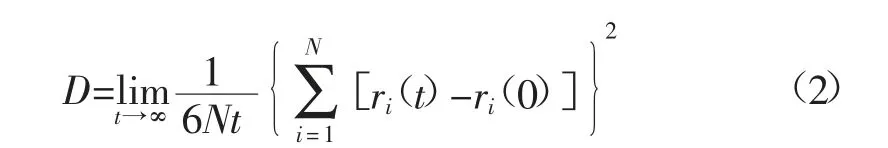
式中:D 为扩散系数;N 为粒子数量,个;t 为时间间隔,s;ri(t)为分子在t 时刻的质心位置,nm;ri(0)为分子在0 时刻的质心位置,nm。
由式(2)计算在298.15 K 下可得自由扩散系数为3.550 22×10-11,与文献[10]模拟的数值相接近,与实验数据相差稍大,原因可能在于在模拟过程中的考虑含氧官能团的影响及与实际孔径有差别。观察其数量级发现其对于吸附的影响并不大,故在吸附过程中可忽略其分子的自由扩散。
2.6 吸附等温线及吸附量
O2在最接近井下温度的298.15 K 和303.15 K下70~120 kPa 的吸附等温线如图12。

图12 常温常压下吸附等温线Fig.12 Adsorption isotherm at normal temperature and pressure
由图12 大致可以得出在同等压力下,吸附量因温度变化而变化,大致趋势是随着温度升高,O2的物理吸附量是降低的,在同等温度下上升趋势大致相同,说明压力对煤吸附量的影响大致呈梯度上升,且呈正相关,大致关系为一次函数关系,煤氧物理吸附量相对来说较小,为简单预测常温常压下的煤氧物理吸附量,分别得出298.15、303.15 K 下70~120 kPa 下的压强与煤氧物理吸附量的数学关系。
在298.15 K 情况下为:

在303.15 K 情况下为:

式中:x 为压强,kPa;y 为吸附量,mmol/g。
在模拟过程中,为研究煤物理吸附O2的最大吸附量,使煤物理吸附O2达到饱和,在温度298.15、303.15 K 下逐步提高压强,吸附饱和时吸附等温线如图13。由图13 可发现,煤物理吸附O2的最大吸附量是非常大的,在压力从50 MPa 往后曲线的上升的缓慢,说明煤的物理吸附O2逐渐趋近于饱和,在压力达到近100 MPa 时煤物理吸附O2达到饱和,且在温度一定时,随着压强的增加,煤物理吸附O2量逐渐上升,且不容易达到饱和,直至填满孔隙后才达到饱和。
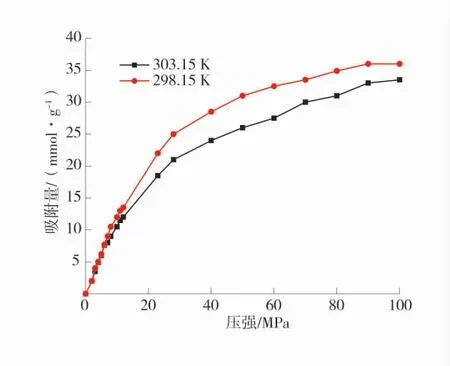
图13 吸附饱和时吸附等温线Fig.13 Adsorption isotherm at adsorption saturation
为了更加细致研究温度变化对吸附量的影响,在等压情况下进行了温度变化的模拟,等压情况下10~90 ℃的吸附量如图14。可知:在同等压力下随着温度升高,煤的物理吸附量总体呈下降的趋势,在10~40 ℃之间的下降趋势变化较快,在60~90 ℃之间下降稍缓慢,在40~60 ℃之间有些波动,原因可能与煤升温后煤的孔隙度变化有关。与实验的变化规律一致,在10~100 ℃的范围内会出现波动,且与煤的孔隙度变化有关。为更加准确的估算,以温度为横坐标,吸附量为纵坐标,得出在100 kPa 下温度与煤物理吸附O2量的数学关系式为:y=-0.015 7x+0.253 8。故此式可用来估计在常温常压下的煤物理吸附的量,是对于煤氧吸附机理的补充细化。

图14 等压情况下10~90 ℃的吸附量Fig.14 Adsorption capacity at 10 - 90 ℃under constant pressure
3 结 论
1)温度与O2物理吸附量呈负相关,在100 kPa时其线性回归方程为y=-0.015 7x+0.253 8;压力与吸附量呈正相关,且在常温常压下均可得出其回归直线方程,用来预测O2的物理吸附量;温度与等量吸附热呈负相关,压力对等量吸附热的影响相对来说较小,在实际计算中可以忽略不计;随着温度增加吸附能量的最大值在0 附近,且分布概率的最大值随温度增加而增加,意味着相互作用逐渐减弱,吸附量下降;以密度分布图的形式验证上述的结论,结合能量分布、吸附量、等量吸附热等因素得到更加系统的煤物理吸附O2的特性,为后续研究煤自燃机理奠定了理论基础。
2)温度为298.15 K 时的O2的自由扩散系数为3.550 22×10-11m2/s。在吸附过程中可忽略其分子的自由扩散。
3)O2与含氧官能团的吸附作用影响分别为吡咯>苯>萘>芘>菲>
先吸附位点为羟基,对于抑制煤物理吸附氧气作用机理的细化补充提供参考。
猜你喜欢
数学小灵通(1-2年级)(2024年2期)2024-05-14 09:22:58
航空材料学报(2023年6期)2023-12-18 05:23:50
文萃报·周五版(2023年47期)2023-12-03 09:44:45
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27 09:22:44
数学小灵通(1-2年级)(2022年12期)2022-12-23 05:38:16
中国音乐学(2022年1期)2022-05-05 06:48:46
科普童话·学霸日记(2021年12期)2021-05-30 15:44:23
奇妙博物馆(2021年4期)2021-05-04 08:59:48
小演奏家(2018年9期)2018-12-06 08:42:02
物理学报(2018年10期)2018-06-14 06:31:32
