
Differential analysis of intestinal microbiota and metabolites in mice with dextran sulfate sodium-induced colitis
2022-11-25 08:11:56JiaLiWangXiaoHanJunXiangLiRuiShiLeiLeiLiuKaiWangYuTingLiaoHuiJiangYangZhangJunCongHuLiMingZhangLeiShi
World Journal of Gastroenterology 2022年43期
Jia-Li Wang, Xiao Han, Jun-Xiang Li, Rui Shi, Lei-Lei Liu, Kai Wang, Yu-Ting Liao, Hui Jiang, Yang Zhang,Jun-Cong Hu, Li-Ming Zhang, Lei Shi
Abstract
Key Words: Ulcerative colitis; Gut microbiota; Metabolites; Dextran sulfate sodium; Mucin
INTRODUCTION
Ulcerative colitis (UC) is a chronic nonspecific inflammatory disease of the colorectum and has an unknown cause. UC is more likely to occur in young and middle-aged people. Its main clinical symptoms include diarrhea, mucopurulent bloody stools, and a variety of extraintestinal manifestations,such as peripheral arthritis, erythema nodosum, anterior uveitis, and others that seriously affect the quality of life of patients. The World Health Organization lists UC as one of the most difficult modern diseases and as one of the most significant research topics in the field of digestion because it is difficult to cure, easily recurs, has a high risk of cancer, and often requires lifelong medication.
UC pathogenesis is complex and considered to be mediated by genetic susceptibility, microbial dysregulation, and environmental factors[1]. The intestinal barrier is a complex and prominent defense system that not only prevents the transfer of various toxins through the intestines into tissues and organs but also prevents the invasion of endogenous microorganisms, which is particularly prominent in maintaining homeostasis in the intestine and even the body[2]. The intestinal barrier consists of a mechanical, chemical, biological, and immune barrier. The role of biological barriers in UC is receiving increasing attention. The latest research shows that abnormalities of the intestinal microbiota and its metabolites play a more important role in the progression of the course of UC[3]. In addition to being involved in constituting a biological barrier, the intestinal microbiota and metabolites also have the functions of biological antagonism, defense against infection, participation in the maturation of the immune system, regulation of intestinal mucus, regulation of intestinal epithelial metabolism, and nutrition[3].
Studies have confirmed that the number of goblet cells in the intestine of aseptic reared mice decreases, their morphology becomes smaller, the mucus layer becomes thinner, and the permeability increases compared with conventionally reared mice[4]. Jakobssonet al[5]found that in C57BL/6J mice with two different intestinal microbiotas, although the thickness of the colonic mucus was similar, there was a significant difference in the permeability of the mucus barrier. Several other studies have found that intestinal microbiota and metabolites can affect mucus through the following effects: (1) Regulation of mucus synthesis: the microbiota's structures, such as lipopolysaccharides, flagella proteins, and inflammatory factors induced by induction, can directly regulate the mucus 2 protein, Mucin2 (MUC2)to produce mucus; probiotics can also provide the relevant nutrients required for MUC2 synthesis to upregulate its transcription; bacterial products and the inflammatory factor secretion caused by them also induce the formation of sulfotransferase, sulfate MUC2, and enhance the anti-inflammatory capacity of the intestine[6,7]; (2) Mucus secretion regulation: active substances, such as lipopolysaccharides and secondary bile salts, stimulate goblet cells to secrete MUC2 into the intestinal cavity to flush bacteria and resist invasion, but excessive MUC2 secretion causes goblet cell depletion and reduces the anti-infection ability of the intestinal mucosa[8,9]. It has also been suggested that inflammasomes can maintain the intestinal environment and microbiota homeostasis by promoting the production of interleukin-18 in sensing intestinal bacterial products or cell damage[10]; and (3) Mucus degradation regulation: primarily the slow degradation of probiotics represented byAkkermansia muciniphila, which is used to maintain the dynamic balance of the mucus layer and the rapid degradation ofEscherichia coli, a representative of pathogenic bacteria that destroys the integrity of the mucus layer.
Therefore, the intestinal microbiota and its metabolites can directly constitute a biological barrier and affect the mucus barrier to play a multi-dimensional regulatory role. It can be seen that the intestinal microbiota and metabolites have a central position in the pathogenesis of UC. At present, although many studies have confirmed the important role of intestinal microbiota in the pathogenesis of UC, the intricate connection between microbiota and metabolites in UC occurrence remains unclear, and the correlation analysis of UC differential microbiota and its metabolites is relatively scarce, suggesting the need to carry out further studies.
Based on the intestinal microbiota and metabolism, this study uses a combination of antibiotics to establish pseudo-aseptic mice. We followed the internationally accepted dextran sulfate sodium method to establish a UC model. By comparing the severity of the disease, mucus-related protein expression,and 16s rDNA sequencing of pseudo-aseptic and bacterial UC mice, we evaluated the practical significance of intestinal microbiota in UC pathogenesis in multiple dimensions. Further, through microbiota sequencing combined with non-targeted metabolomics association analysis, we tried to reveal the differential intestinal microbiota and microbiota metabolites in UC pathogenesis, exploring more sensitive biomarker compositions and providing new ideas for diagnosing UC.
MATERIALS AND METHODS
Experimental animals
The animal protocol was designed to minimize pain or discomfort to the animals. Forty male C57BL/6J mice, specific pathogen free (SPF) grade, weighing 20 ± 2 g were purchased from SPF Biotechnology Co., Ltd. (Beijing, China, certificate No. SCXK-2019-0010). They were routinely raised in the SPF-level animal room of Beijing University of Chinese Medicine, with a 12-h light/night cycle, temperature of 22± 2 °C, and humidity of 50%-60% and were given access to unlimited food and drinking water. The experimental process complied with the ethical requirements formulated by the Laboratory Animal Ethics Subcommittee of the Academic Committee of Beijing University of Chinese Medicine (No.BUCM-2020-01162). The study design was shown in Figure 1.
Establishment of pseudo-aseptic models
Amphotericin B (catalog No. 1397-89-3), vancomycin hydrochloride (catalog No. 1404-93-9), neomycin sulfate (No. 1405-10-3), metronidazole (No. 443-48-1), and ampicillin sodium (No. 69-52-3) were all purchased from China Shanghai Macklin Biochemical Co., Ltd. After 1 wk of adaptive rearing of all mice, 40 mice were randomly divided into the pseudo-aseptic group (N group,n= 15) and the nonintervention group (K group,n= 25) using the random number table method. A schematic diagram of the model to replicate the pseudo-aseptic model of N group mice is shown in Figure 2A. Gastric lavage(0.1 mL/10 g) was first given with amphotericin B (1 mg/kg body weight) for 3 d, and from day 4, 1 mg/mL of ampicillin was added to drinking water. Meanwhile, mice were given a mixture of antibiotics that included vancomycin 50 mg/kg body weight, neomycin 100 mg/kg body weight, metronidazole 100 mg/kg body weight, and amphotericin B 1 mg/kg body weight every 12 h for a total of 21 d (i.e., 4-24 d)[11].
Establishment of a colitis model
Pseudo-aseptic mice were labeled as pseudo-aseptic colitis model groups (group A,n= 15), and nonintervention mice were randomly divided into the following two groups using the random number table method: Blank group (group F,n= 10) and bacterial colitis model group (group G,n= 15). In addition to the blank group, the remaining 30 mice were free to drink 2.5% (w/v) dextran sulfate sodium (DSS) solution (molecular weight: 36000-50000 Da; MP Biomedicals, Santa Ana, CA, United States) to establish an acute experimental colitis model.
Analysis of the disease activity index
The disease activity index (DAI) score includes the following three indicators: weight loss rate, stool consistency, and degree of fecal occult blood. The DAI score = (weight loss rate score + stool consistency score + fecal occult blood score)/3[12], the specific scoring criteria are shown in Table 1.
Measurement of colorectal length
The abdominal cavity was exposed, and the pelvis was cut from the anus with scissors. The fascia and other structures from the rectum were cut with forceps and ophthalmic scissors upwards. An ileocecal mass, a large bulge, was discovered, cutting from the lower end of the ileocecal to the rectum, and the colorectal length was measured.
Measurement of colorectal wet weight
The removed colorectal wet weight was measured before dipping in the phosphate buffer solution, and the intestinal tube was cut along the longitudinal axis before weighing. The feces in it were cleaned up,and the wet weight of the intestinal tube was recorded.
Colonic histological lesions score
After the specimen was fixed with 4% paraformaldehyde for 24 h, 4 μm sections were prepared after paraffin embedding. The histological lesions of the colon were observed under light microscopy after hematoxylin-eosin (HE) staining. The standard histological lesions scoring was used[13]. Table 2 provides the scores, with a total score of 0-16. One slice was made per mouse, and the average score of seven items per specimen was calculated.

Table 1 Disease activity index assessment standards
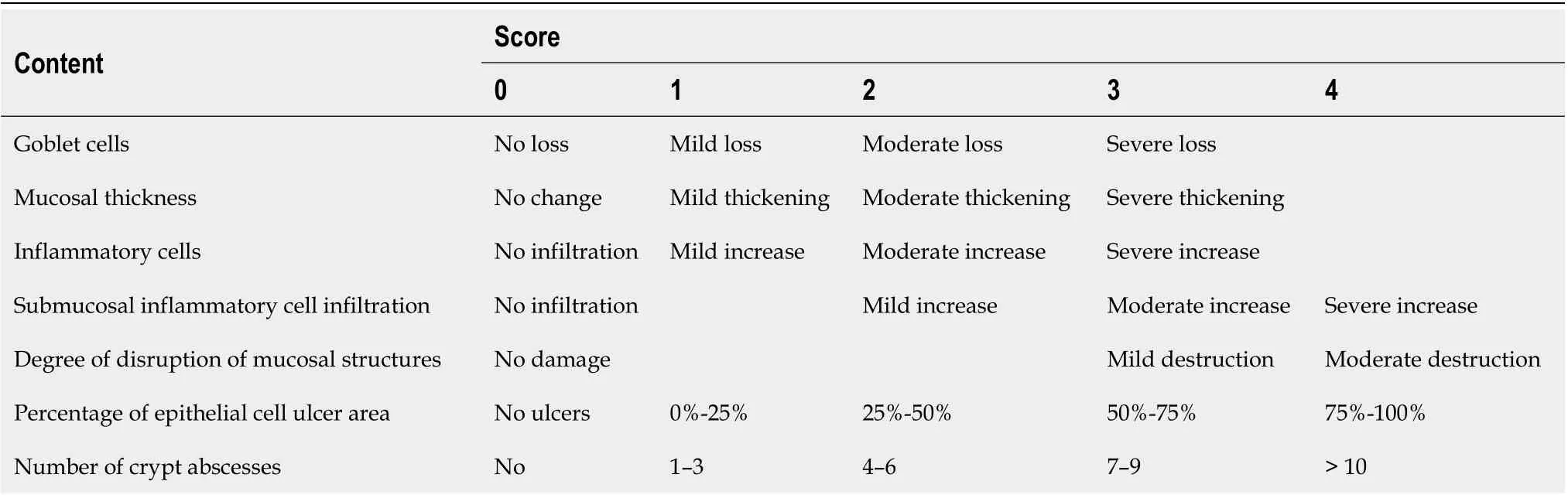
Table 2 Scoring criteria for histological lesions in the colon
Colon tissue mucopolysaccharide alcian blue staining
The paraffin sections were stained with alcian blue dye, and the HE staining step was continued. This was followed by washing, counterstaining, dehydrating, and clearing post-sealing. A panoramic scanner was used to scan the specimen, and the Caseviewer software (Hungarian 3D HISTECH,Pannoramic 250/MIDI, version 2) took pictures. Each sample was photographed after setting the white balance of the high magnification field of view (× 400), and Image J software (National Institutes of Health, United States) was used to analyze the images. The area of positive staining was selected for statistical analysis.
Colonic MUC2 immunofluorescence staining
Paraffin sections were de-waxed and then repaired by antigens. Primary antibodies (MUC2, 1:200,GB11344, Wuhan Servicebio Biotechnology Co., Ltd., Wuhan China), secondary incubated antibodies,and 4′,6-diamidino-2-phenylindole-stained nuclei were added after fluorescence quenching and serum blocking. Each sample was photographed and collected after setting a high magnification field of view(× 200) white balance; the image was analyzed using the Image J software mentioned above. The percentage of the positively stained area (area%) was selected for statistical analysis.
16s rDNA sequencing of the intestinal microbiota
The genomic DNA of the sample was extracted using a DNA extraction kit, followed by agarose gel electrophoresis. NanoDrop2000 was used to detect the concentration of the DNA. Takara's Tks Gflex DNA Polymerase was used for polymerase chain reaction (PCR), ensuring amplification efficiency and accuracy using genomic DNA as a template and specific primers with barcodes. Identification of the microbiota diversity corresponding V3-V4 regions was done using primers 343F and 798R. This study sample was assayed for mouse fecal microbiota, and the V3-V4 zone forward primer was set as 343F-5′-TACGGRAGGCAGCAG-3′, and the reverse primers used were: 798R-5′- AGGGTATCTAATCCT-3′. The PCR products were detected by electrophoresis, purified with magnetic beads after detection, purified as a two-round PCR template, amplified by two-round PCR, and then used for electrophoresis detection, purification with magnetic beads after detection, and Qubit quantification of the PCR products after purification. The PCR products were mixed in equal amounts according to PCR product concentrations and sequenced on a PCR machine (580BR10905; Bio-Rad Laboratories, Hercules, CA,United States). Using Vsearch (version 2.4.2) software, the high-quality sequence valid tags obtained by quality control were operational taxonomic unit (OTU) classified according to 97% similarity. The sequence with the largest abundance in each OTU was selected as the representative sequence of the OTU. The Ribosomal Database Project classifier naive Bayesian classification algorithm was used to compare the representative sequence with the database to obtain the OTU annotation information.
Aseptic conditions were used to collect mouse feces. The feces were retained on clean filter paper,immediately picked up with disinfected forceps, placed in cryopreservation tubes, frozen in liquid nitrogen, and stored in a -80 °C ultra-low temperature freezer for subsequent use in the sequencing of fecal microbiota. The biological information for differential microbiota compared on a website (https://cloud.oebiotech.cn/task/) included the following: OTU abundance, community distribution statistics of microbiota, alpha diversity analysis, beta diversity analysis, and microbial multivariate statistical analysis [differential species heat map and 16S-based Kyoto Encyclopedia of Genes and Genomes (KEGG) function prediction].
Gas chromatography-mass spectrometry analysis and identification of intestinal microbiota metabolites
Gas chromatography-mass spectrometry (GC-MS) was used to detect and identify metabolites of the fecal microbiota. Processes included: sample pretreatment, metabolite extraction, metabolite derivatization, GC-MS detection, data pretreatment, and statistical analysis. The chromatographic conditions of this study were: DB-5MS capillary column (30 m × 0.25 mm × 0.25 μm, Agilent J&W Scientific, Folsom,CA, United States), high purity helium carrier gas (purity not less than 99.999%), flow rate 1.0 mL/min,and a temperature of 260 °C at the inlet. The injection volume was 1 μL, the injection was not shunted,and the solvent was delayed by 5 min. Program heating was as follows: The initial temperature of the column oven was 60 °C, maintained for 0.5 min, heated to 125 °C at a rate of 8 °C/min, heated up to 210°C at a rate of 5 °C/min, heated up to 270 °C at a rate of 10 °C/min, and heated up to 305 °C at a rate of 20 °C/min for 5 min. Mass spectrometry conditions: Electron bombardment ion source, ion source temperature of 230 °C, four-stage rod temperature of 150 °C, and electron energy of 70 eV. The scanning method was full scan mode, and the quality of scanning ranged from m/z 50-500. Metabolite data analysis included multivariate statistical analysis, univariate statistical analysis, differential metabolite screening, correlation analysis, and metabolic pathway enrichment analysis.
Statistical analysis
SPSS 22.0 software (SPSS Inc., Chicago, IBM, United States) was used to analyze all data. Measurements that conform to the normal distribution were expressed as mean ± SD, witht-test analysis for comparisons between two groups and one-way ANOVA for multi-group comparisons. Fischer’s least significant difference was used for homogeneous variances and Tamhane's T2 for unequal variances.The measurement data of the abnormal distribution were expressed as median and quartile spacing(P25, P75), with the Mann-Whitney nonparametric test used for comparison between two groups and the Kruskal-Wallis nonparametric test used for multi-group comparison. The intergroup comparison of the counting data was expressed using median and interquartile spacing (P25, P75), and the Kruskal-Wallis nonparametric test was used for multi-group comparisons of the counting data. The statistical methods of the present study were reviewed by Prof. Zhao-Lan Liu from the Center for Evidence-based Chinese Medicine, Beijing University of Chinese Medicine.
RESULTS
Combination of five antibiotics could effectively establish an intestinal pseudo-aseptic mouse model
After 24 d of combined antibiotic application, there was no significant difference in body weight between pseudo-aseptic mice and mice in the non-intervention group (25.52vs25.64,P> 0.05,Figure 2B). The number of OTUs in the intestinal microbiota of non-intervention mice was significantly higher than that of pseudo-aseptic mice (2259.50vs389.83,P< 0.01, Figure 2C), indicating that the abundance of intestinal microbiota in pseudo-aseptic mice was significantly reduced. Subsequently, a columnar accumulation of species relative abundance at the phylum taxonomic level was plotted,showing the top 15 species in abundance, and it was found that the intestines of non-intervention mice were dominated by theBacteroidetes(77.1%) andFirmicutesphyla (18.8%). In contrast, the intestinal communities of pseudo-aseptic mice were dominated by theProteobacteriaphylum (96.9%), with a distinctly single community structure (Figure 2D). A further selection of the Shannon index for the violin plot showed that the species richness and microbiota community diversity of the intestinal microbiota of the non-intervention mice were significantly greater than those of the pseudo-aseptic group (6.75vs1.78,P< 0.01, Figure 2E). The Rank Abundance analysis found that the span of the horizontal axis of the non-intervention mice curve was significantly larger than that of the pseudoaseptic mice, indicating that the composition of the non-intervention mouse microbiota was relatively rich, and the span of the longitudinal axis of the non-intervention mice curve was smaller than that of the pseudo-aseptic mice, indicating that the species composition of the non-intervention mice microbiota was more uniform (Figure 2F). Further beta diversity analysis found that the two groups of microbiota differed substantially between groups and could be significantly separated (Figure 2G).Based on the above results, it was confirmed that the combined antibiotic method adopted by this study could effectively remove the intestinal microbiota of mice and establish an intestinal pseudo-aseptic mouse model, and its microbiota abundance, community structure, and alpha and beta diversity are significantly reduced.
Mice with bacteria were more severely ill in DSS-induced colitis
On day 2 of taking the DSS solution, stool changes and fecal occult blood test were positive, and the mice gradually lost weight from day 3 onwards. The symptoms gradually worsened with the prolongation of the DSS drinking time. During the entire molding period, four mice died in the pseudoaseptic group and five mice died in the bacterial group. Performing DAI scoring on the last day of molding revealed an increase in both groups of colitis mice and greater severity of illness in the bacterial mice (Figure 3A). After dissecting the intestinal tube, the length of the colorectum and the wet weight of the intestinal tube were measured, and it was found that the intestines of the two groups of colitis mice were significantly shortened and that the wet weight was significantly increased, especially in the bacterial mice. (Figures 3B-D). Subsequently, 10 mice in each group were selected for histological lesion evaluation of the intestinal mucosa, and the results showed that the four layers of colonic mucosa structure in the blank group were clear, the goblet cells were abundant, there was no obvious inflammatory cell infiltration, and the mucosal thickness was normal. In contrast, the mucosal structure of the colitis mice was destroyed; the four-layer structure disappeared, the inflammatory cells infiltrated, the submucosal layer was partially affected, and no obvious crypt abscess was seen (Figure 3E). The histological performance of pseudo-aseptic mice was between blank and bacterial mice, and the histological lesions were light. The score was significantly lower than that of bacterial mice (Figure 3F).
Intestinal mucus loss was more pronounced in DSS-induced colitis mice with bacteria
HE staining revealed that goblet cells in the intestinal mucosa of colitis mice were reduced. Then,through alcian blue staining, it was found that the colon mucosa of blank mice was rich in mucopolysaccharides and goblet cells. The mucopolysaccharides and goblet cells in the colons of mice with colitis were significantly reduced, the mucus layer was interrupted, and the goblet cells were significantly reduced and vacuolated (Figures 4A and B), and the characteristics of mucus of pseudo-aseptic mice came in between those of blank and bacterial mice. Immunofluorescence staining of MUC2, the most important structural and functional mucus protein, was significantly decreased (P <0.01) in the intestines of the bacterial colitis model compared with those of the blank group. Expression of MUC2 in the pseudo-aseptic colitis mice came in between that of the blank and bacterial mice (Figures 4C and D).
The above two studies suggest that different intestinal microbiota have different effects on mucin and mucus, and the degree of colitis that eventually forms is different. The method of free drinking DSS solution can successfully establish a colitis model, but the symptoms of colitis, disease activity, intestinal mucosal histological structure, and mucus distribution in pseudo-aseptic mice are less than those in intestinal bacterial mice. The appearance of this difference may be related to the diversity of the intestinal microbiota or the difference in microbiota metabolites between pseudo-aseptic and bacterial colitis mice. The study then used 16s rDNA sequencing combined with non-targeted metabolomics technology to reveal this characteristic difference, further explore the potential differential intestinal microbiota and metabolites of colitis mice, and screen out the characteristic differential microbiota and metabolites in colitis.
Analysis of different characteristic microbiota in DSS-induced colitis mice
Six mice were randomly selected from each group by the random number table method for 16s rDNA microbiota sequencing, and by comparing the number of OTUs, it was found that the OTU number of the intestinal microbiota of blank group mice was significantly higher than that of mice with bacteria (P< 0.01, Figure 5A). Subsequently, the diversity index dilution curve with goods coverage index and specaccum species accumulation curves were constructed, and the results showed that the two curves tended to be stable at the end, indicating that the sequencing data of each sample were sufficient and that sampling was sufficient (Figures 5B and C). The observed species index was used to plot the box plot and found that the alpha diversity of the intestinal microbiota of mice with bacterial colitis was significantly lower than that of the blank group (Figure 5D). Beta diversity analysis of the composition of the intestinal microbiota of the two groups of mice was further studied, and the results showed that the two groups were separated farther apart, which proved that there was a significant difference(Figure 5E).
Subsequently, Wilcoxon rank sum test was used for clustering at the genus taxon level of the microbiota, and Wilcoxon was performed on the different species of intestinal microbiota, and 50 different species withP(FDR) < 0.05 and names were selected for heat map cluster analysis (Figure 5F).Compared with that in the blank group, it was found that the following bacterial genera in the intestinal microbiota of mice with bacterial colitis were significantly increased (P< 0.05):Anaerobiospirillums,Rikenellaceae RC9gut group,Prevotellaceae Ga6A1group,Escherichia-Shigella,Klebsiella,Anaerotruncus,Negativibacillus,Candidatus Stoquefichus,Blautia,Verrucomicrobium UBA1819,Helicobacter, andLachnoclostridium. The above bacterial genera were significantly elevated in mice with colitis and can be considered harmful bacteria that cause the occurrence or aggravation of colitis.
The bacterial genera that significantly decreased (P< 0.05) in the intestinal microbiota of mice with bacterial colitis were:Bacteroides,Prevotellaceae UCG-001,Prevotellaceae NK3B31,Acetatifactor,Muribaculum,Lactobacillus,Eubacterium coprostanoligenes,Candidatus Saccharimonas, andEubacterium brachy. The above bacterial genera were significantly reduced in colitis mice, and we think that they may be able to prevent or mitigate the occurrence and development of colitis.
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States analysis was then used to predict the composition of differential microbial gene functions of known phylum level classifications, and the predicted KEGG results were displayed using cluster heat maps (Figure 5G). The study found that in the classification of the phylum level there were 28 KEGG signaling pathways with obvious enrichment of different microbiota in mice with colitis. Combined with the characteristics of the colitis disease itself andP(FDR) values, we deduced that the pathways involved were, from large to small according to the difference in significance: biodegradation and metabolism of symbionts, cell growth and death, biosynthesis and metabolism of glycans, energy metabolism, immune system diseases, digestive system, nucleotide metabolism, metabolic diseases, metabolism of terpenoids and polyketone compounds, amino acid metabolism, protein folding, classification and degradation, cancer,metabolism of cofactors and vitamins, immune system, metabolism of other amino acids, transport and catabolism, lipid metabolism, cellular processes and signaling, signaling molecules and interactions, and enzyme families. In summary, it was observed that the functional prediction of differential intestinal microbiota was mainly concentrated in the pathways related to the metabolism of various substances.Thus, we carried out further research on microbiota metabolism.
Analysis of different microbiota metabolites in DSS-induced colitis mice
Orthogonal partial least-squares discrimination analysis was used to distinguish the overall differences in metabolic profiles between groups and look for differential metabolites between groups. It can be found that the microbiota metabolites of the blank group and the colitis mice have good polymerization,the intra-group difference is small, and the separation between the groups is obvious, indicating that there are obvious differences between the two groups of metabolites (Figure 6A). ThePvalues and fold change values of the two groups performed on thet-test were visualized using the volcano plot(Figure 6B), and the differential metabolites between the two groups were further screened (the standard was set to the variable weight value > 1, and thePvalue of thet-test < 0.05). Subsequently, all significantly different metabolites were hierarchically clustered, and a clustered heat map was plotted(Figure 6C). After analysis, it was found that there were 7 kinds of metabolites with significant downregulation in the intestinal microbiota of mice with bacterial colitis, and according to the significant differences, the order was: Galacturonic acid, maltose, lactitol, D-ribose, pyrophosphate,lactulose, and N-acetyl-5-hydroxytryptamine; there were 50 kinds of metabolites significantly upregulated in the intestinal microbiota of mice with bacterial colitis, the top 10 metabolites by the significant difference in size were: D-tagletose, o-phosphateserine, 5-methoxytryptamine, spermine,ribonic acid, creatinine, pinitol, sarcosine, 3-hydroxybutyric acid, and phenol (Table 3). Subsequently,through correlation data matrix analysis (Figure 6E), the degree of correlation between significantly different metabolites was quantified, and it was found that the significantly upregulated differential metabolites in colitis were most closely related to pyrophosphate, lactulose, galacturonic acid, maltose,and lactitol, and the significantly upregulated metabolites were closely related to o-phosphate, sonol, Ltryptophan, butylenediamine, 5-methoxytryptamine, spermine, p-hydroxyphenylpropionic acid, and llactic acid.
Based on the KEGG database, the study used the KEGG ID of differential metabolites to enrich the metabolic pathways of differential metabolites, and theP(FDR) < 0.05 in the metabolic pathway was used as the significance of the enrichment of the metabolic pathway, and the significant enrichment signaling pathway of the top 20 was selected for bubble mapping (Figure 6D). The analysis found that the differential metabolites of colitis mice were mainly enriched in: biosynthesis of aminoacyl tRNA;arginine and proline metabolism; ABC transporter; biosynthesis of valine, leucine, and isoleucine; cAMPsignaling pathway; metabolism of glycine, serine, and threonine; metabolism of alanine, aspartic acid,and glutamic acid; pantothenic acid and CoA biosynthesis; GABA synapses; biosynthesis of phenylalanine, tyrosine, and tryptophan; D-arginine and D-ornithine metabolism, glutathione metabolism,butyrate metabolism, and hypoxia-inducible factor-1 signaling pathways. In general, the differential metabolites of the intestinal microbiota of colitis mice are mainly concentrated in amino acid and energy metabolism.

Table 3 Differential metabolites of gut microbiota in dextran sulfate sodium-colitis mice

1.9350.028-3.003 Diacetone alcohol ↑1.0000.029-0.917 Beta-glutamic acid ↑1.4420.029-1.651 Sinapinic acid ↑1.8740.031-2.768 Atropine ↑1.2110.031-1.903 L-asparagine ↑1.1030.035-0.9884',5-dihydroxy-7-glucosyloxyflavanone ↑1.3340.037-1.861 L-glutamine ↑2.5300.039-4.230 Adenosine ↑1.0700.039-0.948 Pyruvic acid ↑1.0510.041-0.985 Cerotinic acid ↑1.0260.043-0.942 L-tyrosine ↑1.6460.043-2.361 Urocanic acid ↑Up-arrows represent upregulated metabolites, and down-arrows represent downregulated metabolites in microbiota+ colitis mice.
Finally, the correlation analysis of differential metabolites and differential microbiota species of the two groups of intestinal microbiota was carried out using Speedman correlation analysis, and the correlation matrix of the differential metabolites of top 20 and the microbiota at the genus taxonomic level was plotted (Figure 6F). The results showed that 3-deoxyhexanol, ascorbic acid, 2,3-butanediol,trihydroxybutyric acid, L-tryptophan, levo valine, leucine, o-phosphoserine, pineol, butylamine, and sarcosine were not only positively correlated with the genera significantly elevated in colitis (P< 0.05)but also significantly negatively correlated with the significantly reduced genera (P< 0.05), from which it can be speculated that the above substances are differential metabolites that can cause or aggravate colitis, and are positively correlated with their occurrence and development. Galacturonic acid was positively correlated with the significantly reduced genera in colitis (P< 0.05), while lactulose was negatively correlated with the significantly elevated genera (P< 0.05). Therefore, it can be considered that galacturonic acid and lactulose may play a beneficial role in the occurrence and development of colitis.
DISCUSSION
This study found that bacterial colitis caused more mucosal damage and inflammation than bacteria pseudo-aseptic colitis. The destruction of intestinal mucus was more pronounced, speculating that the intestinal microbiota contributed to colitis. The differences in the microbiota could determine the severity of colitis. However, in the past, the academic community has discussed the mechanism of the intestinal microbiota in UC, mostly based on research on the characteristics of the microbiota itself, such as flagellar protein, lipopolysaccharide,etc., often ignoring the co-metabolism of the host and the microbiota or the self-metabolism of the microbiota. Its metabolites can be used by the host intestinal epithelial cells again, thereby participating in and maintaining the intestinal microbiome balance of the host[14]. Modern research has gradually concluded that the intestinal microbiota in UC can affect the intestinal microenvironment through different characteristics of the species itself and metabolites produced by metabolism, including affecting the function of epithelial structures, changing the state of mucus synthesis, secretion, and degradation, and promoting or inhibiting immune responses and inflammatory responses[15,16].
We found that certain microbiota and metabolites were significantly elevated in colitis mice after comparing them to blank mice, suggesting that the microbiota and metabolites were involved in colitis or aggravated it. Significantly reduced microbiota and metabolites in colitis are deemed to have a protective effect on colitis. Further, the correlation analysis between the differential microbiota and metabolites and the analysis of KEGG pathway enrichment found that the intestinal differential microbiota and its metabolites in mice with colitis were mainly concentrated in amino acid and energy metabolism.
There are 10 to 100 trillion microorganisms in the human gastrointestinal tract, and in the past few decades, the impact of the gut microbiota on human health has received widespread interest from science and the general public. The gut microbiome comprises bacteria, viruses, fungi, and archaea that live in different states in the human gastrointestinal tract. More and more studies have confirmed that the intestinal microbiota is closely related to inflammatory bowel disease, metabolic disease, liver disease, hypertension, and other diseases[17,18]. As probiotics and other means to restore and rebuild the normal intestinal microbiota have shown good therapeutic effects for a variety of diseases, the intestinal microbiota is considered to be a new target for the occurrence or treatment of many diseases[19].
However, because the intestinal microbiota not only has a large number but also has a complex distribution and structural composition, the driving mechanism of the intestinal microbiota in many diseases is still unclear, which hinders the construction of intestinal microbiota models for specific diseases and the search for disease characteristic intestinal microbiota. Therefore, the development of aseptic mice is the most effective tool for conducting intestinal microbiota research, and aseptic mice can provide scientific methods for solving such problems[20].
Currently, two main methods have emerged in academia to study the effects of the microbiota on mouse physiology and disease: aseptic mice and antibiotic treatment regimens (pseudo-aseptic mice).Aseptic mice do not contain any bacteria, viruses, eukaryotic microorganisms, or other saprophytic or parasitic related life forms[21], but the high labor and cost of generating and maintaining them have resulted in many research teams not being able to use this model. In addition, this chronic lack of stimulation from foreign antigens can leave the development of its organs in an idealized aseptic state,leading to significant limitations in the structure and function of organs in the body, especially the immune system[22].
Scientists have created another method using multiple antibiotics for gut sterilization to solve this problem. Broad-spectrum antibiotic therapy usually eliminates most of the intestinal microbiota of mice.It can be easily applied to any genotype or condition in mice. Still, because it is impossible to eliminate the intestinal microbiota, it is generally referred to as pseudo-aseptic mice relative to aseptic mice[23].Different antibiotics selectively deplete different members of the microbiota, with metronidazole and clindamycin targeting anaerobic bacteria; vancomycin being effective only against gram-positive bacteria; polymyxin B specifically targeting gram-negative bacteria[24]; amphotericin B having strong antimicrobial activity against candida spores,etc.; neomycin being effective against both gram-positive and gram-negative bacteria; and ampicillin being more effective against gram-positive bacteria,Viridans streptococciandEnterococcus[25]. Pseudo-aseptic mice can therefore be established by extensively depleting the intestinal microbiota by using different kinds of antibiotic mixtures. In this study, the establishment of intestinal pseudo-aseptic mice was built by combining species antibiotics, and the 16s rDNA sequencing of the microbiota was confirmed.
The pathogenesis of ulcerative colitis is complex, and most of the current views are that the interaction of multiple factors causes it. The academic community summarizes the pathogenesis of UC as follows: in genetically susceptible people, such as those suffering from depression and anxietyrelated conditions, the intestinal microbiota involved in the intestinal barrier is destroyed, and the immune system function is disrupted, resulting in an excessive immune-inflammatory response[26].The pathogenesis of UC involves multiple mechanisms, and the colitis model established by different methods is suitable for different research purposes, among which DSS is the most widely used chemical drug for the preparation of colitis models. In the DSS model, sulfated polysaccharides act as a direct chemical toxin of the colon epithelium by reducing the number of mucins and destroying the intestinal epithelial mucus layer[27], causing intestinal epithelial cells damage and structural destruction of the tightly connected complex, while breaking the distribution of the intrinsic intestinal microbiota and affecting the bacterial community structure and diversity[28]. The permeability of the intestinal barrier is increased, and harmful macromolecular antigens, bacteria, toxins, and other substances successfully pass through the intestinal barrier, causing abnormal responses to inherent and adaptive immunity and cascading amplification of inflammation and eventually forming colitis.
A growing number of epidemiological clues point to the onset of UC being closely related to dysbiosis of the gut microbiota. Colonic contents provide bacteria with a rich nutritional environment and become the main settlement of the intestinal microbiota, which provides energy and nutrients to the host through metabolites[29]. Under physiological circumstances, the intestinal microbiota mainly has the following functions: first, it forms a biological barrier with the intestinal mucosal epithelium, blocks exogenous antigenic substances from entering the intestinal mucosa, and produces different metabolic and secondary metabolites through its metabolism to provide energy for the intestinal epithelium;secondly, it participates in the differentiation of T lymphocytes, and jointly regulates mucosal immunity with lymphatic centers and immune cells to regulate immune function; thirdly, it participates in the metabolism and synthesis of a variety of amino acids, proteins, and other substances and participates in the composition of the mucosal barrier[30].
Studies have shown that bacteria that promote mucus degradation, such asRuminococcusandEntamoeba histolytica,have been found in larger numbers in patients with UC compared to healthy individuals.Listeriathat inhibits mucus synthesis has increased significantly, in contrast,Lactobacillusthat promotes MUC2 secretion has decreased significantly in monocytogenes[31].Escherichia coliinfection can disrupt the balance of intestinal flora and disrupt the intestinal microenvironment[32]. By colonizing the intestinal epithelial cells and secreting enterotoxins,E. colidamages the intestinal mucus layer, reduces the protein expression of tight junctions and adhesive junctions, impairs the intestinal mucosal barrier structure, increases intestinal permeability, and induces inflammatory responses and the production of cytokines, such as tumour necrosis factor-α, in addition to increasing MUC2 degradation, which is directly involved in UC pathogenesis[33]. In the stool specimens of patients with UC, theProteobacteriaphylum increased significantly, and the phylaFirmicutesandBacteroidetesdecreased significantly[34], accompanied by different degrees of expression changes in various amino acids, such as decreased valine and glutamate expression; increased tryptophan and isoleucine content;increased pyruvate metabolism; decreased citric acid expression; and decreased short-chain and medium-chain fatty acids in energy metabolism[35]. Other studies have shown that the protective microbiota ofClostridium,Prevoella,Bifidobacterium, andLactobacillus acidophiluswere significantly reduced in UC[36], while the invasiveEkmanellaandE. coliwere significantly increased. The metabolites of short-chain fatty acids, tryptophan, and bile acids[37]that provide nutrients and energy and protect the epithelium were also significantly reduced[38]. BesidesKlebsiella,Escherichia-Shigella,Bacteroides,Lactobacillus, which are well-known microbiota in colitis, we also foundCandidatus Stoquefichus,Anaerobiospirillum,Muribaculum,Rikenellaceae RC9gut group,Candidatus Saccharimonas,Prevotellaceae Ga6A1group, andNegativibacillus. We believe these are emerging microbiota that induces or protects against colitis.
Some studies have reported a certain relationship between other emerging flora and metabolites and colitis. For example,Faecalibacteriumcan influence goblet cell differentiation, mucin synthesis, and glycosylation in colonic epithelium, thereby regulating the intestinal mucus barrier[39].Lachnospiraceaecan degrade mucins and are major consumers of mucins.Lachnospiraceaeover-deplete MUC2, reduce the thickness of the mucus layer and cause severe inflammation in the colon in DSS-induced UC mice[40].Allobaculum mucolyticum secretes a high amount of mucin O-glycan targeting carbohydrate active enzymes, which enables it to efficiently degrade intestinal mucins, thereby degrading the host's protective mucus layer and disrupting the mucus barrier[41]. Blautia’s excessive concentrations in the body may lead to the elevation of secondary bile acids, such as lithocholic acid and deoxycholic acid,which in turn induce UC[42]. A study observed significant differences in the contents ofBrautiain the fecal and mucosal microbial communities of inflammatory bowel disease (IBD) patients and healthy people[43]. It has also been demonstrated intestinal inflammation in IBD patients is associated with increasedFaecalibaculumrelative abundance[44].
In addition, substantial changes have been evident in colitis, including the marked elevation in 3-deoxyhexanol and o-phosphoserine levels and the reduction in galacturonic acid and other microbiotaderived metabolites. Such findings have not been widely reported with regard to the occurrence and development of colitis. By reviewing the literature, we found that MUCs, as a large and complex class of glycosylated proteins, are characterized by important "mucin domains" composed of a protein core. This core contains proline, threonine, and serine, all three of which are known as rich-PTS sequences.Interestingly, through enrichment analysis, we found that these differential metabolites in this study are primarily enriched in the mucin domain of the rich-PTS sequence synthesis pathways. Levyet al[45]studies have shown that taurine and arginine can further promote the synthesis and expression of MUC2 by modulating the NLRP6 signaling pathway, thereby protecting the intestinal epithelium and mucus barrier. Therefore, we believe that these differential microbiota metabolites in colitis are likely to regulate the composition and function of mucus.
This study found differential intestinal microbiota and their metabolites in colitis. Preliminary studies, such as enrichment analysis, have initially found that these differential microbiota and metabolites are likely to be directly involved and affect the mucus barrier of the intestine. However, this study has shortcomings; namely, the lack of verification links for transplanting differentially expressed intestinal microbiota or metabolites into colitis mice; if an experimental design of microbiota or metabolite transplantation is added, we believe that the scientific nature of this study can be further improved, which is a research question we will evaluate further.
CONCLUSION
Candidatus Stoquefichus, Anaerobiospirillum, Rikenellaceae RC9gut group,Prevotellaceae Ga6A1group andNegativibacillusare potentially emerging flora that induce or aggravate colitis, andCandidatus SaccharimonasandMuribaculumare potentially emerging flora that prevent or alleviate colitis. 3-Deoxyhexitol and o-phosphoserine may cause or aggravate colitis, whereas galacturonic acid may play beneficial roles in colitis alleviation and recovery. The differential metabolites of the flora are mainly enriched in the synthesis-related pathways of the rich-PTS sequence of the mucin MUC2 domain. They can influence the composition and function of mucus by regulating mucin expression, and finally act on the mucus barrier to induce aggravation or reduce colitis prevention.
ARTICLE HIGHLIGHTS
Research background
The role of gut microbiota in ulcerative colitis (UC) cannot be ignored; however, most of current research is only based on the microbiota itself, ignoring microbiota metabolism. Microorganisms can reduce many biologically active substances, such as short-chain fatty acids, which have strong immunomodulatory effects. Modern studies have reported that the destruction of the integrity of the mucus barrier is an early pathological change in UC. Different gut microbiota and their metabolites can influence the intestinal mucus barrier through different pathways, including altering epithelial structure, affecting mucin synthesis, secretion and degradation, and modulating immune responses.
Research motivation
At present, although many studies have confirmed the important role of gut microbiota in UC, the intricate relationship between microbiota and metabolites in UC has not been fully clarified. Association analysis of differential flora and their metabolites are required. It is worthwhile to conduct this study based on the intestinal mucus barrier to further reveal the potential differential biomarkers of UC.
Research objectives
The aim of the present study was to reveal the differential gut microbiota and metabolites that affect mucus in UC pathogenesis. The regulatory effect provides new evidence and new ideas for UC diagnosis.
Research methods
In the present study, based on the intestinal mucus barrier, taking the intestinal flora and metabolism as the breakthrough point, a combination of antibiotics was used to establish pseudo-aseptic mice, and the widely used dextran sodium sulfate was used to establish colitis mice. Disease severity, mucusassociated protein expression, bacterial 16s rDNA sequences, and non-targeted metabolomes of bacterial and bacterial colitis mice were examined. The tract microbiota and metabolites play potentially important roles in UC pathogenesis by affecting the mucus barrier.
Research results
This study found that: (1) The antibiotics combination can effectively remove the intestinal flora of mice,and reduce bacterial abundance, and α and β diversity, alter community structure, and successfully establish pseudo-aseptic mice; (2) Comparing the bacteria-bearing mice with the pseudo-aseptic mice,the bacteria-bearing mice had more severe colitis based on disease activity index, more severe intestinal mucosal damage, and more obvious intestinal mucus loss; (3) In the intestinal flora of colitis mice,Candidatus Stoquefichus,Anaerobiospirillum,Rikenellaceae RC9gut group,Prevotellaceae Ga6A1group, andNegativibacilluswere significantly increased, whileCandidatus SaccharimonasandMuribaculumwere significantly decreased. The significantly up-regulated metabolites in the intestinal flora of colitis mice included 3-deoxyhexitol and ortho-phosphoserine, and the significantly down-regulated metabolites were galacturonic acid,etc.;and (4) Further enrichment analysis found that the above differential metabolites were mainly associated with amino acid and energy metabolism. Spearman correlation analysis found that 3-deoxyhexitol, o-phosphoserine,etc.were positively correlated with the occurrence and development of colitis, and galacturaldehyde was positively correlated with the occurrence and development of colitis. There was a significant positive correlation between galacturonic acid and lactulose and the reduction and colitis recovery.
Research conclusions
Candidatus Stoquefichus, Anaerobiospirillum, Rikenellaceae RC9gut group,Prevotellaceae Ga6A1group, andNegativibacillusare potentially emerging flora that induce or aggravate colitis, andCandidatus SaccharimonasandMuribaculumare potentially emerging flora that prevent or alleviate colitis. 3-Deoxyhexitol and o-phosphoserine may cause or aggravate colitis, while galacturonic acid may play a beneficial role in colitis alleviation and recovery. The differential metabolites of the flora are mainly enriched in the synthesis-related pathways of the rich-PTS sequence of the mucin MUC2 domain. They can affect the composition and function of mucus by regulating the expression of mucin, and finally act on the mucus barrier to induce aggravation or reduce colitis prevention.
Research perspectives
This study identified some less-reported differential gut microbiota and their metabolites in colitis,which could affect UC progression by modulating mucin synthesis, altering mucus status and the mucus barrier. In future studies, we will carry out in-depth transplantation experiments of fecal bacteria and metabolites to further verify the experimental conclusions of this study.
ACKNOWLEDGEMENTS
We thank the Beijing University of Chinese Medicine and other institutions for funding this project, and all the authors for their efforts.
FOOTNOTES
Author contributions:Wang JL and Han X have contributed equally to this work, and they are co-first authors; Shi L and Li JX conceived and designed the study; Wang JL and Han X performed major experimental work; Wang JL and Shi R acquired and analyzed the results and edited the manuscript; Liu LL, Wang K, Liao YT, Jiang H, Zhang Y, Hu JC, and Zhang LM performed the experiments and statistical analyses; Shi L and Li JX revised the manuscript; All authors read and approved the final version of the manuscript.
Supported bythe 13th Five-Year Plan for National Key R&D Program of China, No. 2018YFC1705405; Scientific Research Innovation Team Project of Beijing University of Chinese Medicine, No. 2019-JYB-TD004; New Faculty Startup Fund Program of BUCM, No. 2022-JYB-XJSJJ078; and National Natural Science Foundation of China, No.82004113.
Institutional animal care and use committee statement:The animal study was reviewed and approved by the Animal Ethics Committee of Beijing University of Chinese Medicine. All animal experiments conformed to the internationally accepted principles for the care and use of laboratory animals (certificate No. SCXK-2019-0010, SPF Biotechnology Co., Ltd., Beijing, China; protocol No. BUCM-2020-01162, The Animal Ethics Committee of Beijing University of Chinese Medicine, Beijing, China).
Conflict-of-interest statement:The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflict of interest.
Data sharing statement:All data are available upon reasonable request from LS, b01350@bucm.edu.cn.
ARRIVE guidelines statement:The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORCID number:Jia-Li Wang 0000-0002-2789-5081; Xiao Han 0000-0003-1340-9692; Jun-Xiang Li 0000-0001-7590-9444;Rui Shi 0000-0002-9374-0897; Lei-Lei Liu 0000-0003-4639-5370; Kai Wang 0000-0001-5510-7719; Yu-Ting Liao 0000-0003-1821-048X; Hui Jiang 0000-0003-3755-8975; Yang Zhang 0000-0002-2704-9846; Jun-Cong Hu 0000-0002-2493-5535; Li-Ming Zhang 0000-0003-2039-7124; Lei Shi 0000-0002-7925-5166.
S-Editor:Zhang H
L-Editor:A
P-Editor:Cai YX
 World Journal of Gastroenterology2022年43期
World Journal of Gastroenterology2022年43期
- World Journal of Gastroenterology的其它文章
- Integrity of the editing and publishing process is the basis for improving an academic journal’s Impact Factor
- Upper gastrointestinal endoscopic findings in celiac disease at diagnosis: A multicenter international retrospective study
- Kyoto classification of gastritis: Advances and future perspectives in endoscopic diagnosis of gastritis
- Current status of minimally invasive liver surgery for cancers
- Salvia miltiorrhiza extract may exert an anti-obesity effect in rats with high-fat diet-induced obesity by modulating gut microbiome and lipid metabolism
- Correction to “MicroRNA-596 acts as a tumor suppressor in gastric cancer and is upregulated by promotor demethylation”