
白腐真菌降解麦麸纤维关键基因挖掘
2022-11-23 10:53:56蒋诗雨
河南工业大学学报(自然科学版) 2022年5期
宋 鑫,苏 浩,蒋诗雨,李 力*
1.河南工业大学 粮油食品学院,河南 郑州 450001 2.四子王旗市场监督管理局,内蒙古 乌兰察布 011800
来源广泛且具有抗氧化、调节肠道菌群等功能活性的麦麸膳食纤维属于木质纤维素,由纤维素、半纤维素和木质素构成,复杂的物化构造限制了其作为生态学重要资源的发展和使用[1]。白腐菌中蕴含完整的细胞外木质素降解酶系统,是目前最高效的木质及膳食纤维素降解微生物之一[2]。实验室前期通过筛选得到1株对小麦麸皮具有高改性效率的白腐菌菌株A.polytricha5.584,通过优化后对小麦麸皮木质素的降解率达到68.72%[3-4],需要在分子层面上对其降解麦麸纤维的机制进行深入研究。
转录组是某个群体或细菌在某一生长发育阶段或各种特殊功能境况下转录得到的全部RNA的集合体,可以从总体高度深入研究基因组结构各种功用和基因组构造,阐明单个生物过程和生物过程中的基因分子机制。转录组测序法(RNA-seq)是研究生物功能基因组的重要方式,同时也是基因功能与构造研究的重要基础与出发点[5]。刘家豪[6]对红侧耳菌的转录组进行测序,发现了与玉米秸秆纤维素、半纤维素和木质素降解有关的基因89个,其中70个为上调基因,证明了溶解性多糖单氧酶在玉米秸秆木质纤维素的分解中起着重要作用。
目前,使用野生型白腐真菌对麦麸纤维进行生物改性已经取得了较好的效果,但其降解麦麸木质纤维素的分子机理并不明晰,且白腐真菌以麦麸纤维作为单一碳源时,其胞外纤维素酶系的表达调控机制也鲜有报道,因此难以使用分子生物学方法对其进行定向突变和选育[7]。作者通过RNA-seq对白腐菌A.polytricha5.584在不同碳源下的差异表达基因开展功能注释、GO与KEGG功能分类和蛋白网络互作的分析,筛选出与麦麸纤维降解相关的基因,为在分子水平上揭示白腐真菌降解麦麸纤维的机制奠定理论基础。
1 材料与方法
1.1 材料
麦麸:河南中鹤有限公司;菌株A.Polytricha5.584:中国国家普通微生物学菌种保存管理中心福建省三明真菌研究院,试验前将菌株储存于PDA(Potato Dextrose Agar)培养基上或25 ℃的培养基盒中。
Trizol、RNase-free:赛默飞世尔科技公司;异戊醇、乙醇(75%):天津市科密欧化学试剂有限公司。
PDA培养基(g/100 mL):PDA 3.7,KH2PO40.25,MgSO40.15,VB10.05,土霉素 0.02。
麦麸纤维液态培养基(g/100 mL):麦麸纤维5.0,K2HPO40.1,FeSO40.005,KH2PO40.1,MgSO40.02,CaCl2·2H2O 0.01,(NH4)2SO40.2,MnSO40.002。
葡萄糖液态培养基(g/100 mL):葡萄糖2.0,FeSO40.005,K2HPO40.1,KH2PO40.1,MgSO40.02,CaCl2·2H2O 0.01,(NH4)2SO40.2,MnSO40.002。
1.2 仪器与设备
HWS智能恒温恒湿箱:宁波市东南仪表公司;JP-1500B高速万能粉碎机:永康市久品工贸公司;JM-F50胶体磨:浙江省麦特隆机器有限公司;数显恒温式水浴锅:金坛市佳美仪表公司;DHG-9011A电热恒温干燥盒:上海市精宏实验仪器机械设备公司。
1.3 试验方法
1.3.1 麦麸膳食纤维的制备
麦麸磨碎后过40目筛,将麦麸和蒸馏水按质量比1∶ 10配制,用胶体磨处理25 min;干燥后调节加水量(1∶ 10),95 ℃蒸30 min;降温至50 ℃以下,加入HCl调pH值为5.6,升温至95 ℃;加入淀粉酶(1.5%),搅动30 min;降温至50 ℃以下,加入NaOH调pH值至9.0,搅拌均匀;加入碱性蛋白酶(3%),搅动2 h,升温至100 ℃灭酶;酶解的麦麸用自来水洗涤数遍,直到洗涤液不浑浊;置于烘箱中,60 ℃直至烘干。
1.3.2 菌丝培养及取样
将保藏菌株注射在PDA平板培养基(9 cm)上,25 ℃培养14 d使菌丝完全长满平板,取中央菌丝,分别注射至装有100 mL麦麸纤维液态培养基、葡萄糖液态培养基的250 mL三角瓶中,封口后置于27 ℃、180 r/min的振荡培养箱中。培养7 d后依次取培养液6 mL,室温条件下4 000 r/min离心10 min,去除上清液,菌体用无菌水充分重悬3次,洗涤步骤重复5次以上,直至完全去除培养基杂质,菌体洗涤液干净透明。得到不少于200 mg的活菌体,转入冷冻存管封口,储存于液体氮气罐中。
1.3.3 RNA 提取与转录组测序
用液氮把菌株细胞研磨成粉末,转移至2 mL试管中。加1.5 mL的Trizol,充分摇匀后,室温静置5 min,10 000 r/min离心5 min。向细胞上清液中加入200 μL异戊醇(上清液与异戊醇体积比24∶ 1)和1 mL裂解试剂,10 000 r/min离心10 min,把细胞上清液转至等体积异丙醇的试管中,于-20 ℃的冰箱中冷却1 h。13 600 r/min离心20 min后,用1 mL 75%乙醇结晶上清液,并静置3~5 min。RNA用30~100 μL RNase-free水溶解。采集后的RNA通过NanoDrop系统检测浓度,RNA的结构完整性通过Agilent2100生物分析仪测试,检验无误后,开展菌株的转录组测序。
用mRNA富集法对总RNA进行加工。用含有OligodT的磁珠富集含有polyA尾巴的mRNA,然后再用打断buffer将获得的RNA片段化,用随机的N6引物进行反转录,将人工合成的cDNA二链形成双链DNA。将制备的双链DNA末端补平并5′末端磷酸化,3′端成为凸起1个“A”的粘末端,再接上一条3′端有凸起“T”的鼓泡形连接器。把链接物质经过特异的引物实现PCR扩增。然后将PCR物质经过热改性为单链结构,再用一种桥型引物使单链结构DNA环化,获得单链结构环状DNA文库并上机检测。
转录组测序委托深圳市华大基因技术服务公司,利用高通量测序平台BGI-500,开展paired-end测序。为使后续的组装和分析结果更为可信,使用华大自主开发的过滤软件SOAPnuke(版本:v1.4.0;参数:-l 5 -q 0.5 -n 0.1)进行统计,并使用trimmomatic(版本:v0.36;参数:ILLUMINACLIP:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50)进行过滤,过滤后的Clean reads用于后续的研究和分析。
1.3.4 基因信息的基础分析
数据过滤方式:采用SOAPNuke进行统计,trimmomatic进行过滤,过程包括:除去含有连接的reads(连接污染);除去未知碱基N浓度超过5%的reads;除去较低品质的reads(质量值小于10的碱基占该reads总碱基数的比率超过20%的reads)。
参照基因组的比对方式:首先采用Bowtie2 (版本:v2.2.5;参数:-q-p hred64-sensitive-dp ad 0-gbar 99999999-mp 1,1-np 1-score-min L,0,-0.1-p 16-k 200)[8]将clean reads比对到参考基因组顺序,然后再采用RSEM(版本:v1.2.8)[9]估计基因组和转录本的发表水平。
基因信息De novo组装:首先使用Trinity(版本:v2.0.6;参数:-min_contig_length 150-CPU 8-min_kmer_co v 3-min_glue 3-bfly_opts′-V 5-edge-thr=0.1-stderr′)对clean reads(去除PCR重复以提高组装效率)进行De novo装配,之后再通过Tgicl将已组装的标本进行聚类分析除去数据的冗余,从而得到Unigene[10-11]。Trinity包括3个独立模块:Inchworm、Chrysalis、Butterfly,分别处理了大量reads。Trinity将reads建立为大量独立的de Bruijn图,然后对每个图分别提取全长的转录本水平剪切亚型。De novo的组装品质评价:基于组装结果,通过BUSCO数据库对Trinity组装的转录本进行品质评价,通过与保守基因进行比较,进而说明转录组的组装效果[12]。
编码序列CDS(Coding sequence)的预测:使用Trinity推荐的软件Transdecoder(版本:v3.0.1)识别Unigene中的候选编码区,并首先获得最长的开放阅读框,之后再使用Blast比对Swiss-Prot和Hmmscan搜索Pfam蛋白同源顺序,进而预测编码区。具体步骤:(1)使用 TransDecoder.LongOrfs通过对Unigene.fa进行开放的读码框查找,以获得更长读码框顺序;(2)通过序列相似性用Diamond Blastp把Unigene.fa结果比对在Swiss-Prot数据库系统上,然后再使用Hmmscan对Blast结果在pfam数据库检索;(3)利用TransDecoder.Predict估计Unigene.fa的编码顺序。
微卫星序列标记方法:使用MISA(版本:v1.0;参数:1-12,2-6,3-5,4-5,5-4,6-4 100 150)对Unigene进行检测,使用Primer3 (版本:v2.2.2)对检测到的SSR 进行引物设计[13-14]。
1.3.5 基因表达量与表达量分布的分析
通过Bowtie2 (修订版:v2.2.5;参数:-q-p hred64-sensitive-dpad 0-gbar 99999999-mp 1,1-np 1-score-min L,0,-0.1-p 16-k 200)将clean reads比对到基因组顺序上,并且利用RSEM(修订版:v1.2.8)测算各个样本中的基因组表达水平。根据各个样本的表达量FPKM信息,绘制密度图。
1.3.6 差异基因的筛选与功能注释
DEGseq(参数:差异倍数2倍以上且Q-value≤0.001)方法基于泊松分布,根据Wang等[15]描述的方式,进行了差异基因测定。RNA测序也可被视为随机取样的步骤,每次read在样本中都独立而均匀地采样。在这种假设下,来自整个基因组(转录本)的read数目遵循二项分布(并且近似由泊松分布代替)。通过上述的数据模型,DEGseq给出了一种基于MA-plot的新方式,MA-plot是应用于芯片数据的统计分析工具,让C1和C2显示由2个样品中得到的特定基因的read总量,按照Ci~binomial(ni,pi),i=1,2分布,其中ni代表所有比对上的read总量,pi代表该基因的概率。DEGseq定义M=log2C1-log2C2和A=(log2C1+log2C2)/2,并证明在随机抽样假设下,给定A=a (a是A的观察值)的条件下分布,M遵循近似正态分布。对MA图上的每个基因都进行假设并检验H0: p1=p2与H1: p1p2。然后根据正态分布计算P, 参考Scheid等[16]的方法把P-values修正为Q-values。为增加DEGs的准确率,界定了差异倍数为2倍以上且Q-value≤0.001的基因,并遴选为显著差异表达基因。
使用hmmscan(版本:v3.0)、Blast(版本:v2.2.23)和Blast2GO(版本:v2.5.0)对Trinity组装得到的Unigene与七大功能数据库(NR(Non-Redundant Protein Sequence Database)、NT(Non-Redundant Protein Sequence Database)、Swiss-Prot、KOG(Clusters of orthologous groups for eukaryotic complete genomes)、GO(Gene Ontology)、KEGG(Encyclopedia of Genes and Genomes)、Pfam(Protein Families Database))进行比对注释。当基因功能注释结果不同时,为保证生物学意义,按照NR、Swiss-Prot和KOG的顺序将比对结果作为此基因的注释。
2 结果与分析
2.1 转录组数据与质量控制评估结果
采用BGISEQ-500平台一共测了38.18 Gb数据,重组和去冗余后共得出31 531个Unigene,总长度、平均总长度、n50及GC浓度依次为57 226 025、1 814、2 537 bp和52.42%。通过把Unigene比对到七大功能数据库并加以注解,结果依次有26 646 (NR:84.51%)、5 590(NT:17.73%)、16 359(Swiss-Prot:51.88%)、15 404(KOG:48.85%)、18 475(KEGG:58.59%)、8 638(GO:27.40%)以及20 686(Pfam:65.61%)个Unigene获得功能注释。利用Transdecoder检查出24 404个CDS,同样还检查出1 989个SSR散布于1 536个Unigene中。
转录组测序数据及与参考基因组的比对结果统计见表1。各样品碱基质量值Q30均不小于86.65%。各样品的Clean Reads与参考基因组的比对效率为 86.29%~89.49%,数据利用率较高,表明所选参考基因组能够满足后续分析的需求。

表1 测序数据及比对参考基因组结果统计Table 1 Results statistics of sequencing data and reference genome
2.2 麦麸纤维处理组与葡萄糖对照组差异表达基因与表达量分布分析
使用DEGseq软件进行麦麸纤维组与葡萄糖组之间的差异性基因表达分析,以差异倍数≥2且Q-value≤0.001的基因体作为检验标准,得出了2组样品中间的不同表达基因体集,DEGs(differentially expressed genes)共12 096条,当中上调基因8 214条,下调基因3 882条。使用hmmscan、Blast和Blast2GO软件对所有DEGs在各个数据库系统中执行功能注释的结果如表2所示,可知在各个数据库系统中被注释的DEGs总量有所不同,最后共计31 531个DEGs获得注解,共计2 073个DEGs被所有数据库系统同时注释。

表2 注释的差异表达基因数量统计Table 2 Statistics of annotated differentially expressed genes
对于各种注释的差异表达基因数据,选取了KEGG、GO、NR、NT、Swiss-Prot基因注释数据库作出5维的差异表达基因的Venn图(图1),由各样本FPKM密度分布可以看出麦麸纤维液态培养下与葡萄糖液态培养下RNA的表达量密度曲线分布不同,但组间表达密度分布曲线相互接近,表明分组合理且组间存在差异,样本表达量密度如图2所示。
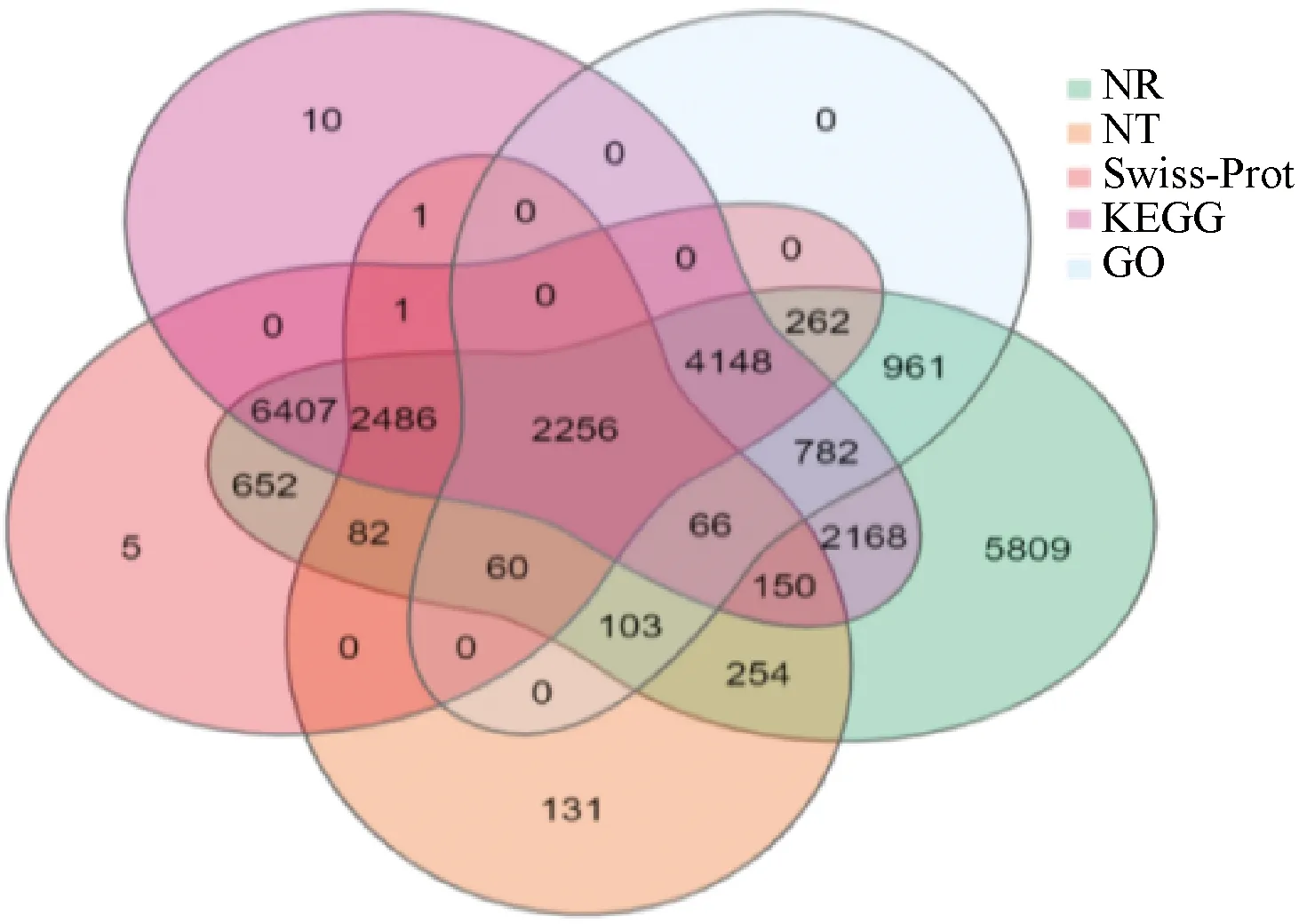
图1 KEGG、GO、NR、NT、Swiss-Prot差异表达基因注释维恩图Fig.1 Venn diagram of differentially expressedgenes by KEGG, GO, NR, NT, and Swiss-Prot

图2 样本表达量密度图Fig.2 Density map of sample expression
GO是一种国际标准化的生物基因活性分类系统,用来描述生命体中基因及其所翻译蛋白质的多种属性[17],并把基因表达形成的不同功能归纳为与生物学过程、细胞组成和分子功能有密切联系的类型。本研究对处理组和对照组通过GO注释进行了统计分析,认为所获得的基因转录组共计8 638个基因得到GO注释,其中有3 415个与得到注释的基因是不同基因,并且它们都被标记在了三大主要功能分类目的42个二级条目中(20+12+10)。在3 415个不同基因中,注释在“生物过程”功能的二级功能“细胞新陈代谢步骤(GO:0008152)”中的不同基因表达数共有1 088个;注释在“细胞组分”功能的二级功能“膜(GO:0016020)”中的差异基因数量有1 111个;注释在“基因分子结构和功能”功能的二级功能“催化活性(GO:0003824)”中的差异基因数量最多,有1 818个。
对全部的3 415个功能差异基因进行GO注释富集解析,筛选出了在三大主要功能分类中校正P<0.01的二级条目共26条,包括“生物过程”类别中的10个类目;“细菌组成”类别中的5个类目;“分子物质构成和功用”类别中的11个类目。综合GO数据库中提出的主要类目功能描述,可将这些类目分为两组:一组是直接参与细胞能量新陈代谢流程,是白腐菌在麦麸纤维等基质环境生长过程中所必需的能量代谢过程;另一组则是参与木质纤维素等麦麸纤维基质氧化分解与代谢过程,包括糖酵解、三羧酸循环、蛋白酶的氧化与还原活性等4个基因本体功能类目,如表3所示。

表3 GO富集麦麸纤维降解相关类目统计Table 3 Statistics of GO enriched wheat bran fiber degradation related categories
KEGG是理解高等各种特殊功能和生物学体系(如细菌、生物学和人类),从分子水平数据资料信息,尤其是大规模分子水平数据信息集形成的基因测序和一些高通量科学实验技术的实用程序数据库系统资源[18],是全球最常见的生物信息数据库管理系统之一。
经过比对,在KEGG信息库中共有4 310条Unigene信息进行了标注,涉及5条主通道:细胞进程(cellular processes)、环境信息处理(environmental information processing)、遗传信息处理(genetic information processing)、新陈代谢(metabolism)、有机合成信息系统(organismal systems);21条子通路[19]。如图3所示,参与新陈代谢总体通路 (global and overview maps)的基因有2 132个;参与新陈代谢下的碳水化合物代谢(carbohydrate metabolism)和细胞进程下的运送和分解代谢物(transport and catabolism)通路的基因分别有1 087和701个;最少的是参与新陈代谢下的外源性生物蛋白与新陈代谢物(xenobiotics biodegradation and metabolism)通路的基因,仅有12个。

图3 KEGG通路分类图Fig.3 KEGG pathway classification map
2.3 蛋白功能网络互作图
为了寻找与麦麸纤维降解过程相关的重要基因,通过分析降解麦麸纤维的白腐菌中不同表达基因,获得相关的调控交互作用网络,并利用在线网站STRING[20]分析不同表达基因的蛋白交互作用网络和Hub基因。使用DIAMOND[21]将所有基因比对至STRING数据库,并使用与所有已知蛋白的同源关系获得所有基因之间的互作关联[22]。基因间的互作关系有评分(150~1 000),得分越高则互作关系越可靠,默认筛选出评分≥300的网络关系作图。
在GO数据库中,将生物调节、碳利用、新陈代谢流程、负控制生物学流程、正控制生物学流程、控制生物学流程、抗氧化活性、结合、催化活性条目下FC(Fold Change)前500的差异基因进行蛋白功能网络互作分析,获得了一个含有不同基因的66条互作关联的蛋白相互作用网络,将互作数据中的Hub基因确认并展示出来,如图4所示。结果显示Hub基因分别为10条互作关系的CL16.Contig1_All和10条互作关系的CL1024.Contig1_All。

图4 GO分类下的蛋白互作网络图Fig.4 Protein interaction network diagram with GO classification
如图5所示,在KEGG条目下,可以从碳水化合物新陈代谢、能源新陈代谢、糖类生物学组成和新陈代谢、辅助因子和维生素代谢通路下FOC值前500的差异基因进行蛋白功能网络互作,获得了一个含有113条互作关联不同基因的蛋白相互作用网络,并将互作数据中的Hub基因确认并展示出来。结果显示,Hub基因分别为10条互作关系的CL16.Contig1_All、CL710.Contig10_All、CL3860.Contig3_All和15条互作关系的CL2915.Contig1_All。它们之间存在较强的相互作用关系,可能是白腐真菌降解麦麸纤维的关键基因,为进一步探索麦麸纤维降解的菌种培养、菌种优化和效率提升提供基础。

图5 KEGG分类下的蛋白互作网络图Fig. 5 Protein interaction network diagramwith KEGG classification
3 结论
通过对麦麸纤维或葡萄糖为单一碳源培养的白腐菌A.polytricha5.584的基因转录组进行高通量测序,获得了12 096个差异表达基因,GO富集结果显示差异表达基因主要参与了白腐菌在麦麸纤维等基质环境生长过程中所必需的能量代谢过程,以及麦麸纤维基质的酶促氧化分解与代谢过程。经GO和KEGG功能注释与富集解析后通过蛋白网络互作分析,得到5个与A.polytricha5.584降解麦麸木质素机制相关的关键基因。其中有1个注释到GO数据库中“氧化还原酶活性”的过氧体水合酶脱氢酶基因(CL16.Contig1_All)表达调高,有2个注释到Pfam数据库中“醛脱氢酶家族”的醛脱氢酶基因(CL1024.Contig1_All、CL2915.Contig1_All),1个注释到Pfam数据库中“短链脱氢酶”的短链脱氢酶基因(CL710.Contig10_All),1个注释到KEGG数据库中“碳代谢”的三羟基萘还原酶基因(CL3860.Contig3_All)。这些基因与白腐菌A.polytricha5.584降解麦麸纤维过程高度相关。其中注释到GO数据库中“氧化还原酶活性”的过氧体水合酶脱氢酶基因(CL16.Contig1_All)同时出现在了经筛选GO条目和经筛选KEGG条目后得到的蛋白网络互作图中,且其在两个蛋白网络互作图中的热度之和最高,值得进一步研究关注。
猜你喜欢
新民周刊(2022年27期)2022-08-01 07:04:49
今日农业(2021年11期)2021-08-13 08:53:24
农家参谋(2021年6期)2021-08-03 03:23:27
传染病信息(2021年6期)2021-02-12 01:52:58
农村农业农民·B版(2019年8期)2019-10-08 05:46:44
农村.农业.农民(2019年16期)2019-01-08 20:34:58
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
化学工业与工程(2015年1期)2015-02-10 03:01:41
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
