
煤基费-托合成α-烯烃水/有机两相氢甲酰化反应的规律
2022-11-12 10:12:20何雨航张安贵张凤申石博文
高校化学工程学报 2022年5期
何雨航, 张安贵, 张凤申, 石博文, 金 欣, 朱 楠
(1. 青岛科技大学 化工学院, 山东 青岛 266042;2. 国家能源集团宁夏煤业有限责任公司, 宁夏 灵武 750411)
1 前 言
煤基费-托合成(Fischer-Tropsch process)是以煤气化合成气为原料,在催化剂作用下将合成气转化为液态烃类化合物的过程[1-5]。通过煤基费-托合成得到的烯烃具有直链α-烯烃含量高、杂质少(基本不含S和N)和碳链分布宽等优点。α-烯烃的重要应用之一是羰基化,即在催化剂作用下,通过合成气对烯烃双键的氢甲酰化反应制备多一个碳的醛,并进一步加氢得到醇[6]。目前,国内增塑剂醇和洗涤剂醇市场对直链高碳醇的需求日益增长,但由于原料成本限制,市场缺口仍很大[7],这为研发利用低成本的费-托α-烯烃氢甲酰化制备高碳醛/醇的新工艺提供了契机。
早期的水/有机两相氢甲酰化体系只局限于C6以下的低碳烯烃[8-9],而针对C6以上的高碳烯烃,利用表面活性剂的乳化和增溶作用,能够有效改善烯烃的传质,进而促进氢甲酰化反应进行。有关阳离子[10-12]、阴离子[13-14]和非离子[15-16]表面活性剂在氢甲酰化反应中的应用被大量报道。目前,基于石油基石脑油开展的氢甲酰化反应的研究也已逐渐受到关注[17-21],然而,含有大量α-烯烃的煤基费-托合成石脑油的氢甲酰化却鲜见报道[22-23],特别是尚未见到有关表面活性剂促进费-托烯烃氢甲酰化的研究报道。
在本研究中,作者以Rh-SulfoXantphos型的水溶性双膦铑为催化剂(图 1,1),SulfoXantphos 为磺化双磷配体2,7-二磺酸基-4,5-双(二苯基膦)-9,9-二甲基氧杂蒽二钠盐,简称SX。C5-C11煤基费-托α-烯烃的水/有机两相氢甲酰化作为模型反应,在阳离子表面活性剂—双十六烷基二甲基溴化铵(DHDMAB)(图1,2)存在下,考察费-托α-烯烃氢甲酰化反应的规律,为水/有机两相条件下费-托α-烯烃氢甲酰化反应的可行性提供实验依据。

图1 本研究应用的铑催化剂和表面活性剂Fig.1 Rh-catalyst and surfactant used
2 实验部分
2.1 试剂与仪器
双(2-乙基己基)琥珀酸酯磺酸钠(AOT,质量分数97%)、双十六烷基二甲基溴化铵(DHDMAB,质量分数97%)、三异辛胺((C8H17)3N,工业级)、乙酰丙酮二羰基铑(Rh(acac)(CO)2,Rh 质量分数40%),北京百灵威科技有限公司;十六烷基三甲基溴化铵(CTAB,质量分数99%)、十四烷基三甲基溴化铵(TTAB,质量分数99%)、十二烷基三甲基溴化铵(DTAB,质量分数99%),上海麦克林生化科技有限公司;壬基酚聚氧乙烯醚(NP-9,质量分数99%),成都艾科达化学试剂有限公司;浓硫酸(H2SO4,分析纯、发烟硫酸(H2SO4·SO3,分析纯)、氢氧化钠(NaOH,分析纯),国药集团化学试剂有限公司;甲醇(CH3OH,分析纯)、无水乙醇(CH3CH2OH,分析纯)、甲苯(C7H8,分析纯),天津市富宇精细化工有限公司;4,5-双二苯基膦-9,9-二甲基氧杂蒽(Xantphos,质量分数97%),北京格林凯默科技有限公司;高纯氢气(H2,体积分数99.999%)、一氧化碳(CO,体积分数99.995%),青岛得一气体有限公司;费-托α-C5、C6和C7烯烃、混合费-托α-C5-7烯烃原料(工业级),宁夏煤业集团有限责任公司;混合费-托α-C5-11烯烃(工业级),上海道普化学有限责任公司;SX 按照文献方法[24]制备。
Bruker AVANCE AV 500 MHz 核磁共振谱仪(NMR,德国Bruker 公司);IRIS INTREPID II XSP 全谱直读电感耦合等离子体发射光谱仪(ICP-AES,美国ThermoFisher 公司);SP-2100A 气相色谱仪(GC,北京北分瑞利分析仪器有限责任公司);气相色谱检测条件:检测器(FID),温度220 ℃;进样口温度220 ℃;载气为高纯氮气,恒流模式50 mL·min-1,分流比30:1;OV-101 非极性毛细管柱(50 m×0.25 mm×0.33 μm);程序升温如下:初始温度80 ℃,保持2 min,升温速率8 ℃·min-1,升至220 ℃,保持20 min。烯烃的转化率Y、产物醛的选择性Sald、正构醛和异构醛物质的量比nn/ni和转化频率(turnover frequency,TOF)通过GC 分析(内标法和归一化法)结果计算得到,计算式如式(1)~(4)所示:


式中:m1、m2、m3、m4和m5分别为原料中烯烃的质量、产物中烯烃的质量、反应生成异构烯烃的质量、正构醛的质量和异构醛的质量,g;x为反应体系中烯烃与铑的物质的量比;t为反应时间,h。
2.2 水溶性Rh-SulfoXantphos 催化剂的制备
采用络合法制备催化剂,具体如下:将1 mg Rh(acac)(CO)2、一定量的SX 和1 mL 除氧去离子水(在氩气存在下,蒸馏去离子水3 次,去除水中溶解的O2)加入60 mL 不锈钢高压磁力搅拌反应釜中,经过合成气(H2与CO 压力比p(H2)/p(CO)= 1/1)置换体系3 次,室温搅拌12 h,然后通入1 MPa 合成气,升温至110 ℃,反应1 h,水浴降温至室温,所得催化剂水溶液无需经过分离即可直接进行下一步氢甲酰化反应。
2.3 Rh-SulfoXantphos 催化费-托α-烯烃水/有机两相氢甲酰化反应的一般过程
向2.2 节新鲜制备的催化剂水溶液中加入定量的表面活性剂和定量的费-托α-烯烃,经过合成气置换体系3 次后充入指定压力的合成气,在指定温度下反应5 h,反应结束后,将反应釜转移至冰水浴中快速降温,泄压开釜,上层有机相经无水硫酸钠干燥后进行GC 分析。
2.4 费-托α-烯烃水/有机两相氢甲酰化体系下Rh-SulfoXantphos 催化剂循环的一般过程
按照2.3 节中的氢甲酰化反应过程,反应结束后经两相分离得到上层包含产物醛的有机相和下层含有铑催化剂的水相,水相在补充新鲜的费-托α-烯烃、表面活性剂和合成气后进行循环实验。
3 实验结果与讨论
3.1 不同的费-托α-烯烃组分含量的分布
费-托α-烯烃通常由α-烯烃和烷烃组成,有时也含有少量的异构化烯烃[6]。研究对宁夏煤业集团有限责任公司提供的费-托α-C5、C6和C7烯烃(由混合费-托α-C5-7烯烃经精馏制备)、混合费-托α-C5-7烯烃和混合费-托α-C5-11烯烃(上海道普化学提供)的组成进行GC 分析,得到每个样品中各种组分的质量分数w(%),以便于对后续氢甲酰化实验的结果进行定量分析。如表1 所示,相较于普通的α-烯烃,费-托α-C5、C6和C7烯烃中均含有一定质量的烷烃,占比为26%~43%,α-烯烃的质量分数均较高,占到52%~67%,异构烯烃仅占3%~7%。对于混合费-托α-C5-7烯烃,总α-烯烃质量分数达到56.6%,其中C6组分质量分数最高(23.3%),其次是C5(18.8%)和C7(14.5%)组分。而混合费-托α-C5-11烯烃中,总α-烯烃质量分数高达70.4%,主要为C6(10.9%)、C7(16.4%)、C8(19.4%)和C9(20.6%),仅含少量的C5(3.0%)及微量的C10(0.1%)、C11(0.01%)组分。综上所述,得到的费-托原料中α-烯烃质量分数较高,异构烯烃质量分数少,便于后续氢甲酰化反应的进行;同时,由于原料中所含的烷烃不参与反应,反应结束后可通过精馏与产物醛实现分离。

表1 不同碳链长度的费托α-烯烃和混合费托α-烯烃的组分含量分布Table 1 Composition distribution of F-T α-olefins and mixed F-T α-olefins with different carbon chain lengths
3.2 Rh-SulfoXantphos 催化剂的31P-NMR 表征
铑催化剂 HRh(CO)2(SulfoXantphos) (图 2,1)由水溶性 SX(图 2,3)与乙酰丙酮二羰基铑Rh(acac)(CO)2在温度θ= 110 ℃、压力p= 1 MPa 的合成气(p(H2)/p(CO) = 1/1)条件下,在水溶液中制备(图2)。得到的铑催化剂水溶液用31P-NMR 进行表征,如图3 所示。作为参比的SX (3)在化学位移δ=-17.7 处出现一个单峰,归属为游离的三价膦的信号(图3(a))。SX 与 Rh 配位形成铑催化剂HRh(CO)2(SulfoXantphos) (1)后,磷原子P 的化学位移向低场移动,并裂分为双峰,出现在δ=27.7 (JRh-P=154 Hz)处(图3(b))。Silva 等[24]的研究也表明,当SX、铑物质的量比较高时(n(SX)/n(Rh) = 10/1),相比于较低的SX、铑物质的量比(n(SX)/n(Rh) = 1/1)时P 的化学位移(δ=22.2)[25],1 中P 的化学位移会进一步向低场移动,出现在δ=25~30 处,这也证实了1 的生成。在δ=15.5 处的单峰能够被归属为3 与Rh(acac)(CO)2形成1 的过程中的中间产物4(图3(b)),在后续氢甲酰化反应进程中,4 会进一步向1 转化。Takahashi等[26]通过X 射线单晶衍射证实了4 的存在。

图2 HRh(CO)2(SulfoXantphos)催化剂的合成Fig.2 Synthesis of HRh(CO)2(SulfoXantphos) catalyst

图3 Rh-SulfoXantphos催化剂的31P-NMR谱图Fig.3 31P-NMR spectra of Rh-SulfoXantphos catalyst
3.3 表面活性剂种类对费-托α-C7 烯烃水/有机两相氢甲酰化反应的影响
在水/有机两相体系下,氢甲酰化反应的速率主要受烯烃的传质控制,C5以上的α-烯烃在水中的溶解度极低,因此本研究以费-托α-C7烯烃作为模型底物考察了添加不同种类的表面活性剂对氢甲酰化反应的影响。如表2 所示,作为空白对比(entry 1),无表面活性剂添加的情况下,α-C7烯烃受到传质限制,导致费-托α-C7烯烃中α-庚烯的转化率、醛选择性和nn/ni均较低,TOF 仅为49 h-1。添加阴离子表面活性剂AOT (entry 2)和非离子表面活性剂NP-9 (entry 3)后,α-庚烯的转化率、醛选择性和正异比均有所提高,但效果并不明显。然而,当季铵盐型阳离子表面活性剂被加入后,催化效率大幅度提升,TOF 值达到400~550 h-1,其中双长链的DHDMAB (entry 7)相比单长链的DTAB (entry 4)、TTAB (entry 5)和CTAB(entry 6),其效果更为显著,α-庚烯基本完全转化,醛选择性达到90% 以上,nn/ni接近50。上述结果能够用Chen 等[27]提出的“胶团-离子对”理论解释:疏水的烯烃易增溶于阳离子表面活性剂在水相形成的正向胶团内,胶团外层的正电层通过库仑力作用富集带负电荷的铑催化剂1,从而促进反应进行,而胶团内的位阻效应也能够有效提高nn/ni值。此外,双长链的DHDMAB 的疏水性更高,临界胶束浓度值更低,对烯烃具有更好的增溶效果。因此,在后续的研究中,均采用DHDMAB 作为模型表面活性剂。
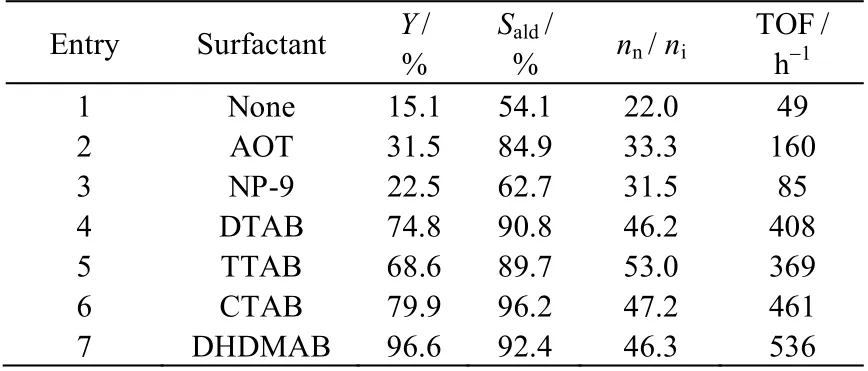
表2 表面活性剂种类对费-托α-C7 烯烃水/有机两相氢甲酰化反应的影响Table 2 Effect of surfactant types on aqueous-organic two-phase hydroformylation of F-T α-C7 olefin
3.4 表面活性剂用量对费-托α-C7 烯烃水/有机两相氢甲酰化反应的影响
如表3 所示为在确保Rh 用量不变的前提下,通过改变DHDMAB 与Rh 的物质的量比,考察表面活性剂用量对氢甲酰化反应的影响。随着DHDMAB 用量的逐渐增大,α-C7烯烃中α-庚烯的转化率、醛选择性、nn/ni和TOF 值先呈现增加的趋势(比较entries 1~3),当n(DHDMAB):n(Rh) = 15:1 时,增加的趋势趋于平缓,而继续增加DHDMAB 的用量,催化性能的改变并不明显(比较entries 3~5)。主要原因是随着表面活性剂用量的增加,催化反应由传质控制逐渐变为动力学控制,反应速率的变化趋于平缓,继续增大表面活性剂的用量反而会导致油/水两相乳化严重,不利于反应结束后油/水两相的分离。因此n(DHDMAB):n(Rh) = 15:1 为最佳的表面活性剂用量。

表3 表面活性剂用量对费-托α-C7 烯烃水/有机两相氢甲酰化反应的影响Table 3 Effects of surfactant dosage on aqueous-organic two-phase hydroformylation of F-T α-C7 olefin
3.5 费-托α-C7 烯烃水/有机两相氢甲酰化反应的动力学
在n(Rh):n(SX):n(DHDMAB):n(α-heptene) =1:10:15:3 000、θ= 110 ℃、p=1 MPa、p(H2)/p(CO)= 1/1 的反应条件下,费-托α-C7烯烃水/有机两相氢甲酰化反应的动力学被研究,通过压降法测定的转化数(turnover number,TON)随反应时间的变化曲线如图4 所示。在最初100 min 内,反应速率较快,α-庚烯的转化近60%,随着时间的延长,由于烯烃的浓度逐渐下降,氢甲酰化的反应速率趋于平缓,当反应时间至300 min 左右时,烯烃基本实现完全转化,因此确定最佳的反应时间为5 h。
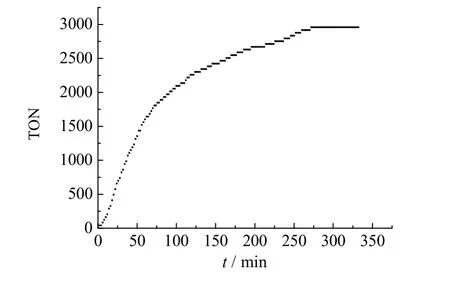
图4 费-托α-C7 烯烃水/有机两相氢甲酰化反应的动力学Fig.4 Kinetics curve of aqueous-organic two-phase hydroformylation of F-T α-C7 olefin
3.6 反应温度对费-托α-C7 烯烃水/有机两相氢甲酰化反应的影响
表4 考察了反应温度对费-托α-C7烯烃水/有机两相氢甲酰化反应的影响。一个明显的规律是,α-庚烯的转化率、醛选择性和nn/ni随着反应温度的升高而增大,当θ= 110 ℃ (entry 4)时,烯烃转化率、醛选择性和nn/ni达到最大值。继续升高反应温度至120℃ (entry 5),由于烯烃异构化副反应速度加快[19],导致醛选择性下降;同时,温度升高更有利于能垒更高的Rh-异构烷烃中间物种的生成[28-29],致使nn/ni也随之降低。因此,确定最佳的反应温度为110 ℃。
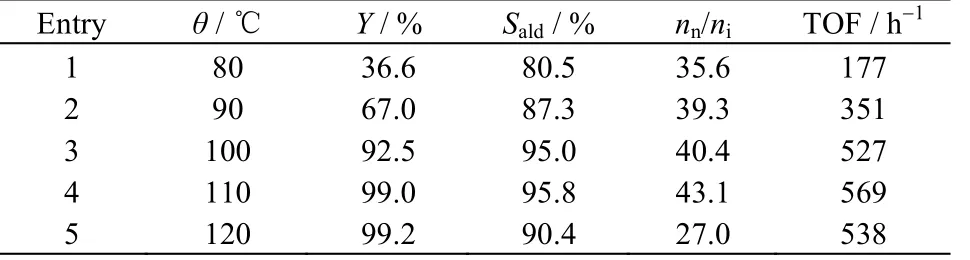
表4 反应温度对费-托α-C7烯烃水/有机两相氢甲酰化反应的影响Table 4 Effects of reaction temperature on aqueous-organic two-phase hydroformylation of F-T α-C7 olefin
3.7 合成气压力对费-托α-C7 烯烃水/有机两相氢甲酰化反应的影响
合成气(p(H2)/p(CO) = 1/1)压力对费-托α-C7烯烃水/有机两相氢甲酰化反应的影响如表5 所示。费-托α-C7烯烃中α-庚烯的转化率随着合成气压力的升高呈现明显的降低趋势,而醛选择性和正异比基本保持不变,这符合氢甲酰化反应的普遍规律[30-32]。主要原因是当CO 的分压过高时,氢甲酰化反应的关键中间体烷酰基铑5 会形成五配位的二羰基烷酰基铑6(见图5),从而抑制了作为速率控制步的H2的氧化加成反应[33]。因此1.0 MPa 的合成气压力被确定为最佳的反应压力。

表5 合成气压力对费-托α-C7 烯烃水/有机两相氢甲酰化反应的影响Table 5 Effects of syngas pressure on aqueous-organic two-phase hydroformylation of F-T α-C7 olefin

图5 CO 压力对催化物种结构的影响Fig.5 Effect of CO pressure on catalytic species structure
3.8 n(SX)/n(Rh)对费-托α-C7 烯烃水/有机两相氢甲酰化反应的影响
SulfoXantphos、铑物质的量比n(SX)/n(Rh)对费-托α-C7烯烃水/有机两相氢甲酰化反应的影响如表6 所示。当n(SX)/n(Rh)在2/1~10/1 变化时,α-庚烯的转化率、醛选择性和nn/ni均保持相对稳定,这与文献报道的规律相符[34-36]。即对于本身空间位阻较大的SX 而言,较低的n(SX)/n(Rh)更有利于催化物种1 的形成。然而,当n(SX)/n(Rh)增至15/1~20/1 时,烯烃转化率和醛选择性呈明显的下降趋势,这归因于在较高n(SX)/n(Rh)的条件下会形成无活性的双螯合物种HRh(SulfoXantphos)2[15],导致烯烃异构化,醛选择性下降。因此,考虑到SX 在催化反应过程中存在氧化消耗,确定最佳的n(SX)/n(Rh)为10/1。

表6 n(SX)/n(Rh)对费-托α-C7 烯烃水/有机两相氢甲酰化反应的影响Table 6 Effects of molar ratio of SX to Rh on aqueous- organic two-phase hydroformylation of F-T α-C7 olefin
3.9 费-托α-C7 烯烃/有机两相氢甲酰化体系下Rh-SulfoXantPhos 催化剂的循环测试
水/有机两相氢甲酰化体系的优点是水溶性膦铑催化剂分配在水相,便于催化剂的分离与循环。表7 考察了在水/有机两相氢甲酰化体系下,在优化的反应条件下,Rh-SulfoXantPhos 催化剂的循环稳定性。在循环过程中,油水两相的分离情况如图6 所示。在首次循环中,铑催化剂表现出的转化率、选择性和nn/ni完全符合优化条件下铑催化剂的催化性能(比较表7 中entry 1 和表6中entry 3)。反应结束后,油水两相能够分相,上层有机相呈透明状,但颜色较深,水相呈乳状液(图6(a)),说明有部分的铑催化剂流失到了有机相。为了验证这一点,将上层有机相减压蒸馏出产物醛后,保留少量残液,然后补加新鲜的费-托α-C7烯烃,再次进行氢甲酰化,α-庚烯的转化率恢复到90% 以上(entry 2)。将反应得到的有机相(图6(b))硝解后进行ICP-AES 分析,证明有14.2% 的Rh 流失到有机相。然而,取第1 次循环后的下层水相进行第2次循环,α-庚烯的转化率下降至43.7% (entry 3),油/水两相能够清晰地分离(图6(c))。显然,水层催化剂活性下降并非Rh 的流失造成的,一个可能的原因是双长链的DHDMAB 具有一定的油溶性,部分流失到有机相,两相传质效率下降,导致催化效率降低。向第2 次循环后的下层水相中补加22 mg (占第1 次用量的67%)的DHDMAB,然后进行第3 次循环(entry 4),Rh-SulfoXantPhos 催化剂的性能再次恢复到第1次循环的水平,从而证明表面活性剂的流失是水相催化剂活性降低的主要原因。第3 次循环后的油/水两相仍能实现有效的分离(图6(d))。在找到循环过程中催化活性流失的主要原因后,作者又重新进行了新一轮的循环实验(entries 5~8)。在每次循环结束后向水相中补加22 mg 的DHDMAB。结果表明,在4 次循环中,烯烃转化率、醛的选择性和nn/ni均保持稳定,TOF 并无较大幅度的下降,总转化数达到11 230,证明铑催化剂具有较好的稳定性。

表7 费-托α-C7 烯烃水/有机两相氢甲酰化体系下Rh-SulfoXantphos 催化剂的循环Table 7 Reuse of Rh-SulfoXantphos catalyst in aqueousorganic two-phase hydroformylation system of F-T α-C7 olefin

图6 铑催化剂分离和循环过程的照片Fig.6 Photos for separation and reuse process of Rh-catalyst
3.10 不同碳链长度的费-托α-烯烃和混合费-托α-烯烃的水/有机两相氢甲酰化
为了考察水/有机两相体系对不同的费-托α-烯烃的普适性,在优化的反应条件下,作者将水/有机两相氢甲酰化体系扩展至费-托α-C5和α-C6烯烃,以及混合费-托α-C5-7和α-C5-11烯烃,结果见表8。如表8 所示,对于α-C5和C6烯烃,该两相催化体系均表现出与α-C7烯烃相当的催化性能,均保持较高的烯烃转化率、醛选择性和nn/ni。进一步扩展至混合费-托α-C5-7和α-C5-11烯烃,结果表明,混合费-托α-烯烃中,各个单一α-烯烃组分的转化率、醛选择性和nn/ni也均能达到较高的水平,从而证明了该催化体系具有良好的普适性。
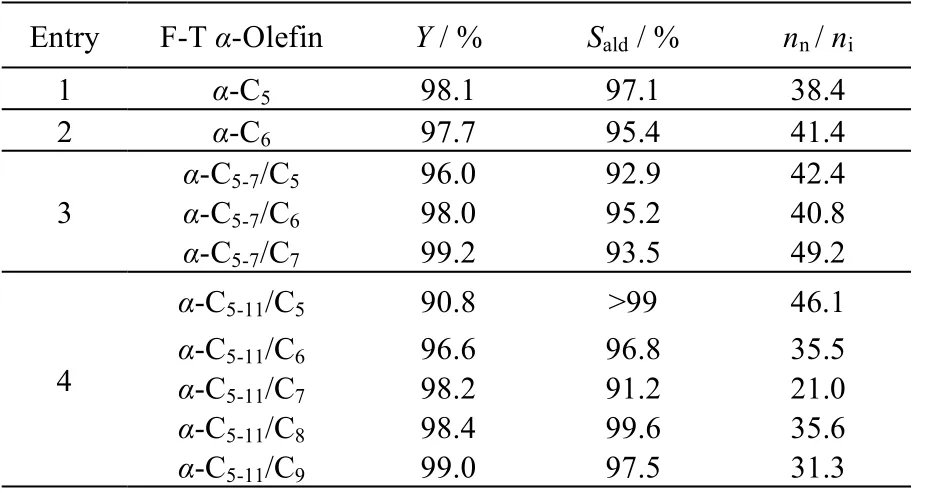
表8 费-托α-C5、C6 和C7 烯烃,混合费-托α-C5-7 和α-C5-11烯烃的水/有机两相氢甲酰化反应Table 8 Aqueous-organic two-phase hydroformylation of F-T α-C5, C6, C7 olefin and mixed F-T α-C5-7 and α-C5-11 olefins
4 结 论
在本研究中,一个表面活性剂促进的高效的水/有机两相催化体系被应用于铑催化的煤基费-托α-烯烃的氢甲酰化反应中。对费-托α-烯烃的GC 分析表明,费-托α-烯烃主要由α-烯烃(质量分数50%~70%)和烷烃(质量分数25%~45%)组成,仅含有少量的异构化烯烃。31P-NMR 证明了磺化双膦配体SulfoXantphos与Rh(acac)(CO)2在合成气作用下形成的HRh(CO)2(SulfoXantphos)催化剂是催化氢甲酰化反应的关键催化物种。通过筛选表面活性剂的种类,确定了双长链的阳离子型DHDMAB 具有最佳的助催化效果。优化了水/有机两相氢甲酰化反应的条件,包括反应温度、合成气压力、n(SX)/n(Rh)、反应时间等。通过将催化体系扩展至不同碳链长度的费-托α-烯烃和混合费-托α-烯烃,证明该催化体系具有良好的普适性。铑催化剂的循环实验表明,通过油/水两相分离,水溶性铑催化剂能够实现循环利用,在补加表面活性剂的条件下,铑催化剂循环4 次后,催化活性和选择性未见明显下降。该研究证明了水/有机两相催化体系在煤基费-托α-烯烃氢甲酰化中应用的可行性,并提供了实验依据。目前,该催化体系的进一步的筛选和优化仍在进行中。
猜你喜欢
分子催化(2022年1期)2022-11-02 07:10:44
云南化工(2021年8期)2021-12-21 06:37:38
中国饲料(2021年17期)2021-11-02 08:15:14
中国石油石化(2021年9期)2021-07-17 09:24:10
中国特种设备安全(2019年5期)2019-07-16 08:52:08
广东饲料(2016年5期)2016-12-01 03:43:22
当代化工研究(2016年2期)2016-03-20 16:21:18
合成化学(2015年10期)2016-01-17 08:56:37
华东理工大学学报(自然科学版)(2015年5期)2015-02-27 13:49:56
华东理工大学学报(自然科学版)(2015年5期)2015-02-27 13:49:56
