
负离子对还原解离反应的影响作用机理
2022-11-11 08:47:28巩颖颖吴延鸿尹昊天周华伟
聊城大学学报(自然科学版) 2022年6期
巩颖颖,吴延鸿,尹昊天,周华伟,徐 振
(1.齐鲁工业大学 化学与化工学院,山东 济南 250399;2.聊城大学 化学化工学院,山东 聊城 252059)
0 引言
近年来,可充电锂离子电池在电化学储能市场占据了主导地位。拥有高能量密度和稳定循环性能的锂离子电池是人们不断追求的目标。其稳定性和安全性与负极表面固体电解液界面膜(solid electrolyte interphase,SEI)的形成和稳定有很重要的关系。SEI膜的形成与生长机理一直是锂离子电池领域受到关注最多的问题。在电池首次充电循环时,电解液组分(包括电解液溶液、锂盐、添加剂)会在负极表面发生还原分解,形成SEI膜,造成电池的不可逆容量的损失[1-3]。形成SEI膜的相关理论研究的重点是电解液组分的构成,成膜添加剂与有机电解液溶剂之间的作用机理也是探究成膜机理的重要方向[4]。
碳酸丙烯酯(PC)以其较高的介电常数(~64)和宽温度范程(熔点-49℃)成为锂电池中常用的低温电解质溶液[5]。PC基电解液存在黏度大(2.513 mPa S)的缺点,在实验中常加入低粘度的线性碳酸酯混合,如碳酸二甲酯(DMC)和碳酸二乙酯(DEC)等。最大的问题是无法在负极表面形成有效的SEI膜,与溶剂发生共嵌入石墨负极,造成石墨负极的剥离脱落[6],降低可逆容量进而降低整个电池的循环寿命。最合适的方法是加入少量的成膜添加剂(2%~5%体积分数)促进SEI膜的形成[7,8],提高锂电池的性能。
在最新的研究报道中,很多实验和理论计算都致力于研究适合在负极成膜的添加剂。不饱和碳化合物碳酸亚乙烯酯(VC)和含卤素的有机化合物氟代碳酸乙烯脂(FEC)[8]、三氟代碳酸丙烯酯(TFPC)[9]、4-氯甲基-1,3,2-二恶唑烷-2-氧化物(CMDO)[10]被证明在负极界面形成有效的SEI膜[11],抑制溶剂的分解。Aurbach等人[12]报道,FEC添加剂分解后形成的LiF在SEI膜中占据主导作用。Wang等人[13]研究表明VC还原电势高于溶剂,优先发生还原解离,分子间电子由VC转移到碳酸乙烯脂(EC),催化了EC的解离。一些基于在实验和理论上(密度泛函理论DFT和从头算分子动力学AIMD)也探究了含硫有机化合物包括亚硫酸乙酯(ES)[14]、亚硫酸丙烯酯(PS)[15]和亚硫酸二甲酯(DMS)[16]以及含磷有机化合物三(2,2,2-三氟乙基)亚磷酸酯(TTFP)[17]等多种添加剂对成膜的改善作用。综上所述,尽管已经有很多的理论和实验研究报道,但是SEI成膜机理仍未被完整的解释清晰,一直是锂电池开发和应用中的重要瓶颈。
为了避免仅在真空条件下模拟的缺陷,本文重点考虑了锂盐负离子参与形成SEI膜的作用,最大限度还原了溶剂化锂竞争体系(FEC)Li+的微观环境,解释了分子水平上对界面成膜的系统的作用,通过DFT计算模拟(FEC)Li+(X)(X=PC,,PC-和PC2-)电解液体系之间的竞争关系,进而对比了不同模型解离前后的差别,最终通过在热力学、动力学的角度以及离子之间相互作用程度上解释负离子效应。
1 实验部分
1.1 模拟体系和计算细节
采用密度泛函理论DFT[18],在ωB97XD/6-311++G(d,p)和B3PW91/6-311++G(2df,2p)水平下,所有模型在介电常数为31.0的混合溶剂PC∶DMC=1∶1(v/v)条件下[19]进行优化。优化后在同一计算级别下进行振动分析得到零点能ZPE和吉布斯自由能G,并确认过渡态搜索没有虚频。原子电荷采用了静电势拟合方法CHELPG。所有计算采用Gaussian 09计算软件包[20]和使用考虑泡利Pauling半径的隐式溶剂模型SMD[21]。最低未占据轨道(LUMO),独立梯度模型(IGM)和分子中的原子(AIM)分析图通过Multiwfn 3.7(dev)软件[22]分析再导入Visual Molecular Dynamics(VMD)软件[23]渲染得到。
2 结果与讨论
2.1 (FEC)Li+(X)的前线轨道及轨道贡献分析
为了预测团簇分子的还原解离能力,计算了5种溶剂化锂模型的最低未占据轨道(LUMO)。基于前线轨道理论,LUMO值越低,代表着分子接受电子能力强,即具有较高的还原能力[24]。图1展示了(FEC)Li+(X)5种模型的LUMO轨道,轨道图的绿色代表正相位,蓝色代表负相位。随着(FEC)Li+在溶剂团簇中占的比重的减少,LUMO从(FEC)Li+分散出去,由图1(b,d)可知,距离(FEC)Li+最近的PC溶剂承载了分散LUMO,而图1(e)两个PC分离(FEC)Li+与的团簇的LUMO却将这种分散减轻。图1(c)所示的(FEC)Li+()模型中,离子直接接触的Li+与上的LUMO分布都比其他模型更加明显,说明负离子所在溶剂团簇的位置影响(FEC)Li+的还原能力。
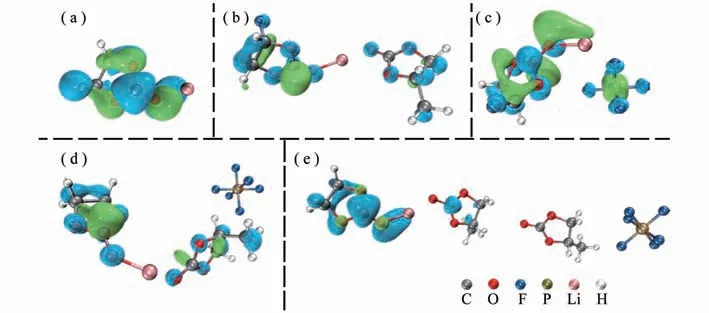
图1 (a)为(FEC)Li+;(b)为(FEC)Li+(PC);(c)为(FEC)Li+();(d)为(FEC)Li+(PC-)以及(e)为(FEC)Li+(PC)2-模型使用SMD-B3PW91/6-311++G(2df,2p)完全优化的LUMO(e V)轨道图(isovalue=0.02),其中绿色蓝色分别为正负相位
为了得到不同模型上LUMO的轨道的空间离域程度,采用Hirshfeld方法计算了轨道离域指数(Orbital delocalization index,ODI)。ODI值越小,说明轨道平摊到越多的原子上,轨道的离域性就越强,在表1中整理和总结了(FEC)Li+(X)的LUMO轨道的ODI和所占LUMO的ODI以及HOMO、LUMO和HOMO-LUMO gap能量值。在(FEC)Li+()的ODI为20.26,低于(FEC)Li+(PC-)和(FEC)Li+(PC)2-的ODI(分别为23.23和23.37),说明在(FEC)Li+()的离域性更强,这与图1中LUMO轨道图分布呈现一致。在整个团簇的ODI中,(FEC)Li+()、(FEC)Li+(PC-)和(FEC)Li+(PC)2-分别为17.06、15.27和15.31,均低于不含的(FEC)Li+(X),说明的存在降低了团簇的ODI,团簇LUMO轨道的离域性增强。其中,(FEC)Li+(PC-)与(FEC)Li+(PC)2-的ODI相近(~15.3),说明两个PC的距离足以削弱了对团簇整体LUMO轨道离域程度的影响。轨道贡献率是对原子轨道贡献值做归一化处理,得到每个原子或者分子片段在整体轨道中的贡献大小。在(FEC)Li+()、(FEC)Li+(PC-)和(FEC)Li+(PC)2-的LUMO轨道贡献分别为18.79%、4.34%和0.12%,可知在(FEC)Li+(PC)2-模型由于两个PC的分隔,对于LUMO轨道的贡献值很低,可忽略其贡献,(FEC)Li+(PC-)模型仍能够产生约4%的轨道贡献。LUMO数值上的参与大约升高了0.04 eV,使得还原能力略微下降。
表1 (FEC)Li+(X)在LUMO轨道的整体及的轨道离域指数(ODI),的轨道贡献率(Contribution of )以及HOMO,LUMO和gap数值,能量单位为eV
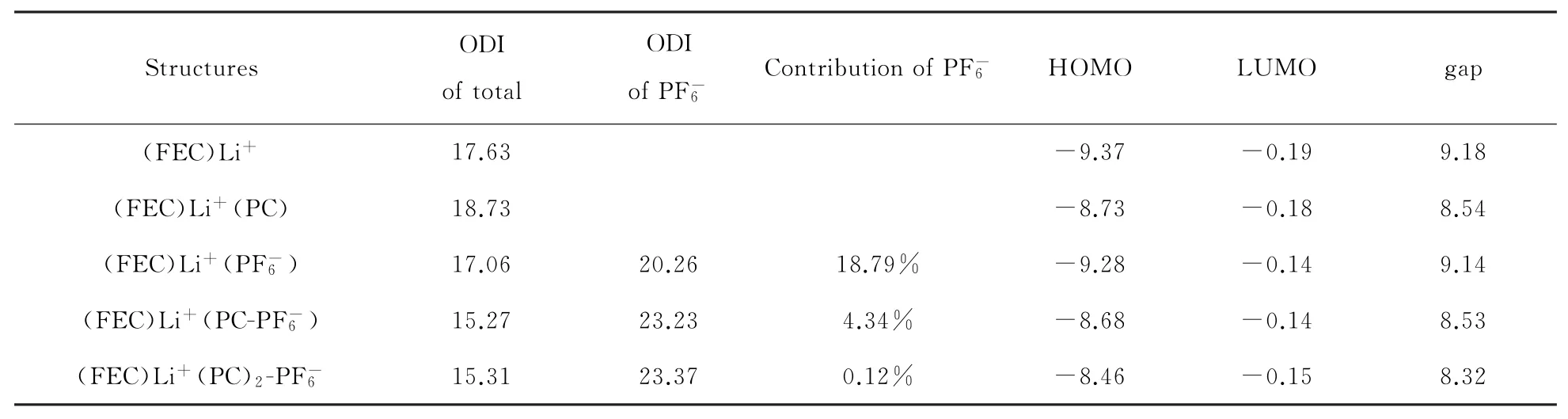
表1 (FEC)Li+(X)在LUMO轨道的整体及的轨道离域指数(ODI),的轨道贡献率(Contribution of )以及HOMO,LUMO和gap数值,能量单位为eV
Structures ODI of total ODI of images/BZ_56_782_1758_854_1796.png Contribution of images/BZ_56_782_1758_854_1796.png HOMO LUMO gap(FEC)Li+ 17.63 -9.37 -0.19 9.18(FEC)Li+(PC) 18.73 -8.73 -0.18 8.54(FEC)Li+(images/BZ_56_782_1758_854_1796.png) 17.06 20.26 18.79% -9.28 -0.14 9.14(FEC)Li+(PC-images/BZ_56_782_1758_854_1796.png) 15.27 23.23 4.34% -8.68 -0.14 8.53(FEC)Li+(PC)2-images/BZ_56_782_1758_854_1796.png 15.31 23.37 0.12% -8.46 -0.15 8.32
2.2 不同理论方法下(FEC)Li+(X)还原分解热力学与动力学分析
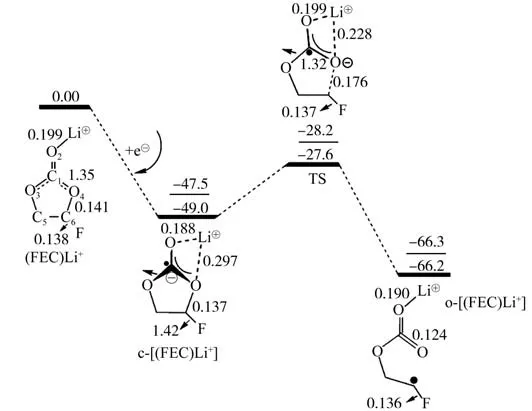
图2 (FEC)Li+分别在ωB97XD/6-311++G(d,p)和B3PW91/6-311++G(2df,2p)(下划线数据)水平下完全优化的单电子还原解离路径的吉布斯自由能示意图,能量以kcal/mol为单位,键长以nm为单位
通过比较两种泛函下的还原解离能垒ΔE a(零点能校正的电子能量)和还原电势φvs Li+/Li,发现B3PW91/6-311++G(2df,2p)高基组水平下展示出更加精准的能量数据。根据表2中B3PW91泛函下的能量变化(/后面的数据),(FEC)Li+(PC-)表现出最低的ΔE a(18.2 kcal/mol),(FEC)Li+与(FEC)Li+()的ΔE a约为19 kcal/mol,从整体上看,5种模型的ΔE a差距在0.9 kcal/mol,说明对解离造成翻越能垒的能力并不明显。还原电势φ的计算由溶剂条件下(FEC)Li+(X)还原到c-[(FEC)Li+(X)]的吉布斯自由能求解得到的,公式如
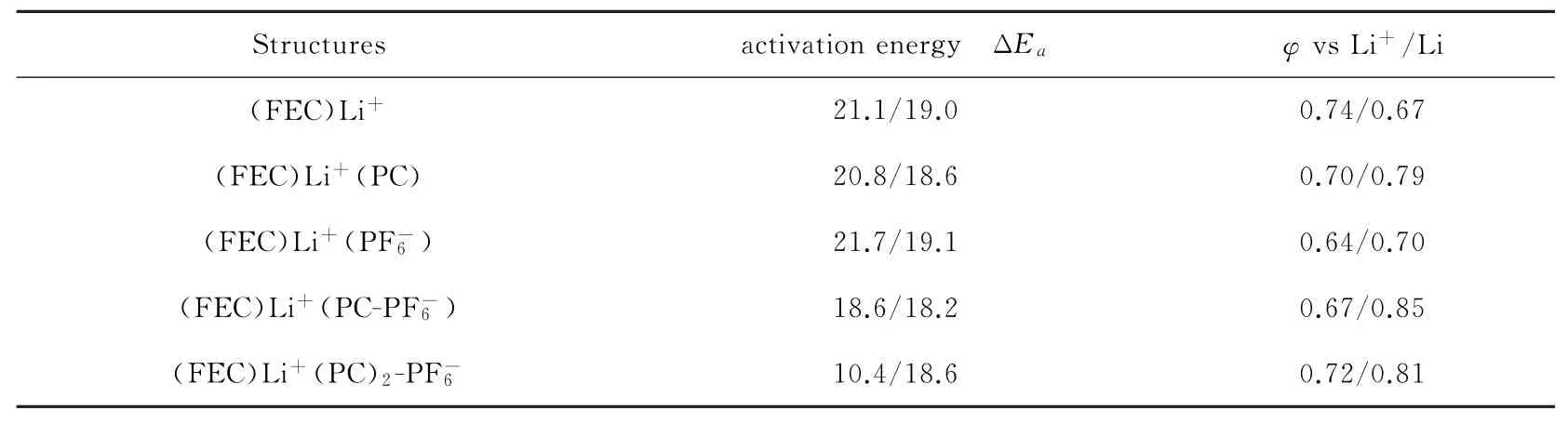
表2 ωB97XD/6-311++G(d),(p)和B3PW91/6-311++G(2df,2p)(/后数据)水平下(FEC)Li+(X)的活化能(ΔE a,kcal/mol)以及解离还原电势(φvs Li+/Li,V)
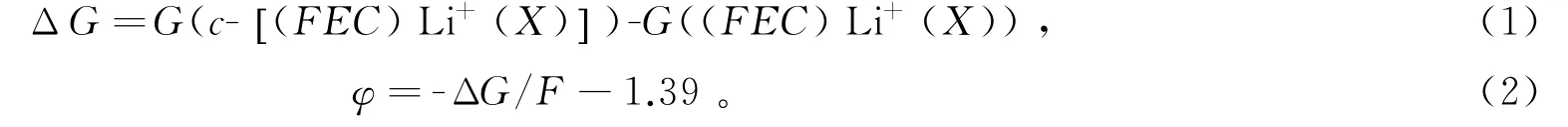
结果表明,Li+与的直接接触模型(FEC)Li+()的还原电势φvs Li+/Li是最接近实验上测得FEC还原电位的0.70 V,常用的计算模型(FEC)Li+为0.67 V略低于实验中的还原电位,说明考虑负离子很好的修正了实验和计算数据上的误差。此外,发现L Li+与共享一个PC溶剂的模型(FEC)Li+(PC-)达到了最高的还原电位(0.85 V),而在Li+与被两个PC分隔的模型(FEC)Li+(PC)2-表达出来的还原电位(0.81 V)与没有的模型(FEC)Li+(PC)的还原电位(0.79 V)接近,说明负离子所带来的还原电位影响因为两个PC的分隔消失了。
2.3 负离子对(FEC)Li+(X)解离前后弱相互作用分析
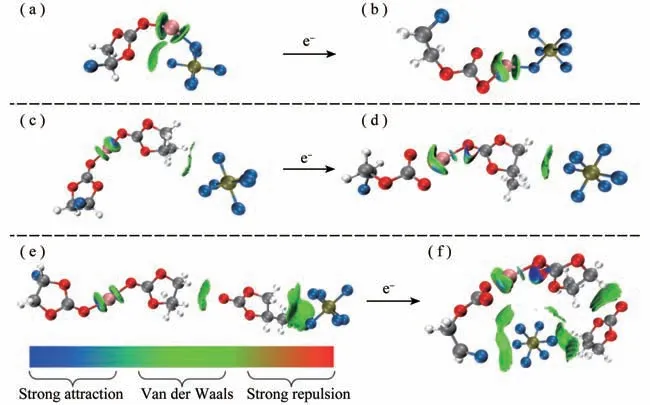
图3 (FEC)Li+()解离前(a)解离后(b),(FEC)Li+(PC-)解离前(c)解离后(d)以及(FEC)Li+(PC)2-解离前(e)解离后(f)的IGM图(isovalue=0.001)
根据IGM初步分析出离子间相互作用的强度和类型,为了获得更详细的具体哪些原子间相互作用,通过搜索键临界点BCP展现键径进行分子中的原子(AIM)分析。图4所示,桔黄、黄、绿色分别代表的是电子密度(3,-1)、(3,+1)和(3,+3)的临界点,桔黄色的曲线对应的是键径。发现的F离子与体系的其他组分都有相互作用,尤其是图4(f)中的5个F离子都在解离后直接与FEC开环阴离子自由基发生作用,明显的键径出现在FEC的F、O与的F-上,这直观的展现了对(FEC)Li+(PC)2-电解液体系在图3中出现大面积的绿色范德华吸引。而图4(b,d)中,解离反应后的的F离子仅有1-2个F与邻近的Li+和PC产生键径,并没有与FEC发生直接的相互作用。图3(a,b)和图4(a,b)说明了(FEC)Li+()解离前的F与FEC的O存在明显的范德华作用,解离后不与FEC直接作用,而是Li+与的一个F依靠静电吸附和范德华吸引共同作用。充分说明了的不同位置影响了电解液体系(FEC)Li+(X)解离还原后的竞争溶剂化结果。
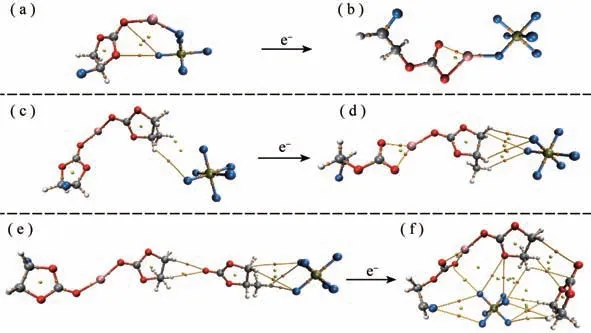
图4 (FEC)Li+()解离前(a)解离后(b),(FEC)Li+(PC-)解离前(c)解离后(d)以及(FEC)Li+(PC)2-解离前(e)解离后(f)的AIM图
3 结论
在使用隐式溶剂模型SMD考虑溶剂效应的条件下,通过密度泛函理论DFT系统的对负离子与Li+直接接触,与Li+共享一个PC溶剂,与Li+被两个PC分隔的模型对比中分别作了系统的理论研究。结果表明参与的FEC的开环解离路径依然与经典路径(FEC)Li+一致,在前线轨道贡献、热力学与动力学和弱相互作用上则表现出差异。整体上的参与,LUMO数值升高了0.04 eV,使得还原能力略微下降。(FEC)Li+()的LUMO轨道分散程度最大,占到轨道贡献的18.79%,其还原电位也是最接近实验中测试的0.70 V,解离后Li+与的一个F依靠静电吸附和范德华吸引共同作用。(FEC)Li+(PC-)的还原电位最高达到0.85 V,解离后主要与PC上的甲基发生范德华吸引。(FEC)Li+(PC)2-对LUMO轨道的贡献接近0.12%可忽略,同时对还原电位的影响也回到0.81 V与不含负离子的(FEC)Li+(PC)(0.79 V)很接近,解离后在微观分子形态上却由链状转变成环状,使FEC的F、O直接与的F通过范德华吸引相互作用。在(FEC)Li+(X)的还原解离过程中,位于电解液体系的不同位置造成明显差异的竞争溶剂化结果。为以后寻找其他PC基成膜添加剂的模型设计在考虑负离子研究方向上做出一方面的研究指导。
猜你喜欢
电子产品可靠性与环境试验(2023年5期)2023-06-04 16:35:39
现代农村科技(2023年3期)2023-04-14 07:02:50
浙江林业科技(2022年1期)2022-02-20 08:36:04
科学技术创新(2022年36期)2022-02-13 04:29:09
科学技术创新(2022年36期)2022-02-01 10:22:22
现代农业科技(2022年1期)2022-01-17 07:17:32
核科学与工程(2021年4期)2022-01-12 06:29:32
科学与生活(2021年3期)2021-11-10 02:29:11
中国机械工程(2019年7期)2019-04-23 07:14:32
钻井液与完井液(2018年5期)2018-02-13 01:06:52
