
硅藻土抑制瓦斯爆炸的微观机理分析
2022-11-11 08:43:24何文浩郝朝瑜张亚超邓存宝
煤炭学报 2022年10期
何文浩,郝朝瑜,张亚超,邓存宝,张 俭
(1.太原理工大学 安全与应急管理工程学院,山西 太原 030024;2.中煤科工西安研究院(集团)有限公司,陕西 西安 710077)
随着我国矿井进入深部开采阶段,瓦斯相关灾害(煤与瓦斯突出、瓦斯窒息、瓦斯爆炸等)越来越难以治理和预防,相关文献统计指出瓦斯相关事故是名副其实的矿井“第一杀手”[1-3]。而其中危害最大的是瓦斯爆炸事故,瓦斯爆炸事故破环性大和死亡率高,始终是矿井安全管理工作的重点,而抑爆减灾技术则是瓦斯爆炸防治工作中的重点内容。
受益于计算机仿真技术的发展,众多学者对甲烷氧化链式反应进行了细致且精确的分析[4-5],并结合敏感性分析对反应机理简化研究[6-8],大幅降低了瓦斯爆炸抑制理论的难度。自此,引起了各类材料对甲烷爆炸影响的理论研究热潮。以常见的水对瓦斯爆炸影响为例。最初,陆守香等[9]指出水作为惰性第三体参与自由基的碰撞反应,以物理方式降低自由基浓度,从而抑制瓦斯爆炸。唐建军[10]通过仿真和试验结合指出:水不仅有物理抑制作用,还能在一定程度与自由基发生反应,降低自由基浓度从而抑制爆炸。文献[11]通过结合量子化学从头算方法和分子动力学模拟表明:水仅在甲烷氧化链式反应初期有抑制作用,而随着链式反应进行到完全激活时,反而会促进甲烷的氧化过程。随着对反应本质的更深入的剖析,水对瓦斯爆炸的影响才得以渐渐清晰。
硅藻土作为一种绿色环保,无毒害的抑爆材料[12],其抑爆效果研究目前主要以试验方式开展[13-15],对具体的微观反应机理则不甚明了。试验方法来测定抑制效果固然清楚,但难以说明内在抑制机理,不便设计改进方案来增强效果。笔者拟从微观角度分析硅藻土对瓦斯爆炸的影响,用量化方法表明影响效果,并指出改进方向。
1 计算方法
本文所有反应物、过渡态和产物结构均在B3LYP-D3/6-311G**水平上进行优化,并在同计算级别下开展频率计算检验和内禀反应坐标(IRC)验证过渡态正确性。所有反应物和产物均不存在虚频,过渡态均只有一个虚频且联通合理反应物和产物。考虑上述计算级别对能量计算的误差,本文所有能量计算均在M062X-D3/Def2-TZVP水平进行。此外,在针对HCHO的吸附能计算中,考虑基组的不完备性对弱相互作用体系影响甚大,对其进行基组重叠误差(BSSE)校正。
1.1 硅藻土模型
硅藻土作为无定形二氧化硅,对其结构的研究较多。文献[16]建立的无定形二氧化硅理论模型指出,其表面具有3种类型羟基:孤立羟基、连生羟基以及双生羟基,该模型很好地解释了硅藻土吸附、催化等各种应用的理论问题。以此理论模型为基础,无定形二氧化硅的计算化学建模主要分为2种方法:第1种为以DFT理论为代表的小簇模型[17-19];第2种是以第一性原理为代表的晶面模型[20]。
笔者综合考虑计算的时长和结果的精度,采用小簇模型来模拟硅藻土表面结构,同时参考关于国产硅藻土结构研究的相关文献[21],选择其中2种常见的硅醇:孤立硅醇(Single Silanol)和连生硅醇(Vicinal Silanol)。模型分子结构如图1所示,孤立硅醇模型如图1(a)所示[22],连生硅醇模型如图1(b)所示[19](分子结构图由可视化软件CYLview导出[23])。
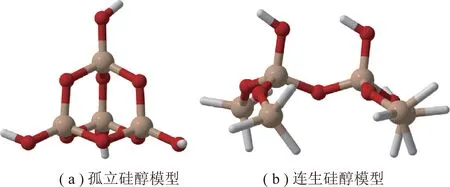
图1 模型分子结构Fig.1 Molecular structure of models
1.2 硅藻土对瓦斯爆炸影响机理分析
1.2.1 与自由基的反应机理
根据甲烷氧化链式反应机理,反应初期自由基浓度对链式反应能否传递下去有着直接关系[24],而在众多自由基中主要以·H,·OH和·HCO为关键链传播自由基[7,25-26]。其中,·HCO由稳定中间产物HCHO与·H,·OH或其他夺H自由基反应后得到[7,24]。如果能降低·H和·OH数量,则·HCO的数量也会大幅降低。
综上,笔者着眼于分析硅藻土对以下2个初始链式反应的影响。
CH4与·H的反应(亦即表2中的:Ori_H反应):

CH4与·OH的反应(亦即表2中的:Ori_OH反应):
若硅藻土与·H和·OH的反应速率能影响甚至超过上述反应,即能有效抑制瓦斯爆炸。由于涉及反应较多,表1为所有可能反应的基本信息。

表1 反应基本信息
表1中反应1~12为硅醇与自由基可能发生的反应。以反应11,12为例,2者均为连生醇与·OH可能发生的反应,反应位点F为图1连生硅醇模型中左侧(B为右侧硅醇),产物分别为H2O和H2O2(略去产生的硅醇自由基),其余反应同理。反应1~4为孤立硅醇与自由基反应,其中反应位点S即代表孤立硅醇。
Reverse反应为反应9产生的连生硅醇自由基与·H的结合反应。
在确定反应过渡态并验证其正确性后,进行相关物质的热力学量计算。考虑甲烷氧化链式反应过程中会引起环境温度上升进而影响热力学量,笔者采用热力学分析软件Shermo进行扫描[27],设定温度为25~1 000 ℃(即298.15~1 273.15 K)[28],每25 ℃计算1次,压力恒定为1 bar(即100 kPa)。由于过渡态频率计算结果中存在较多过小频率(100 cm-1以下),为提高热力学量计算精度,计算模式选用Grimme的准RRHO模型[29]。考虑理论方法的误差,热力学频率校正因子取sclZEP =0.981[30]。
通过热力学量进一步计算得出反应自由能能垒(计算方法见式(1)),结合过渡态理论(TST)计算出各反应随温度变化的速率常数[31],比较速率常数的大小来判断反应相互之间影响大小。
自由能能垒计算公式为
ΔG(T)=GTS(T)-Grea(T)
(1)
式中,GTS(T)为随温度变化的过渡态自由能,kJ/mol;Grea(T)为随温度变化的反应物自由能,kJ/mol。
过渡态理论反应速率常数kTSK计算公式为
(2)
式中,κ为隧道效应校正系数;kB为玻尔兹曼常数,取1.381×10-23J/K;T为绝对温度,K;h为普朗克常数,取6.626×10-34J·s;R为理想气体常数,取8.314 47 J/(mol·K);P0为气相标准态压力;ΔG0为气相标准态压力时,过渡态的自由能与反应物的自由能之差,kJ/mol。
由于本文反应涉及氢转移较多,而该类反应有着较强的隧道效应(特别是低温时),固对其展开细致计算。
隧道效应校正系数κ计算公式有如下2种情况:
(1)近似的Skodje-Truhlar方法(β<α时可用,对应虚频小、温度高时)。
(3)
(4)
(5)
式中,Px为虚频,cm-1。
(2)完整的Skodje-Truhlar方法(本文用于虚频超过1 000的反应)。
当β<α时:
(6)
当α<β时:
(7)
式中,Z为正向势垒,过渡态减反应物的内能,kJ/mol;V为吸热反应为产物减反应物内能,kJ/mol,放热反应取0;NA为阿伏伽德罗常数,取6.02×10-23。
1.2.2 对甲醛的吸附机理
众多研究表明:在甲烷氧化链式反应中,甲醛是不可忽视的关键中间体。甲醛的进一步氧化不仅会大幅加速链式反应的进程,同时还会释放大量的热量来加速反应的进行[4,10,25]。硅藻土由于发达的微孔结构、丰富的表面极性羟基和低廉的成本,常用作极性有机小分子脱除剂[32-33],而甲醛正是典型的极性小分子有机物。
反应物分子总是以极性的方式相互接近,进而达到吸附稳态或发生反应。笔者利用波函数分析软件Multiwfn[34],计算分子表面静电势,最终通过分子可视化程序VMD呈现并得出可能吸附位点[35]。弱相互作用可视化分析选用IRI方法[36],同样通过VMD呈现。
计算弱相互作用时,考虑基组的不完备性,还应加入基组重叠误差(BSSE)的校正。
考虑基组重叠误差(BSSE)的吸附能计算公式如下:
E′Binding=EBinding+EBSSE
(8)
EBinding=Ea+b-Ea-Eb
(9)
式中,E′Binding为校正后吸附能,kJ/mol;EBinding为未校正吸附能,kJ/mol;EBSSE为基组重叠误差校正能,kJ/mol;Ea+b为达到吸附稳态后体系总能量,kJ/mol;Ea,Eb为a,b两分子单独稳定结构时的能量,kJ/mol。
2 结果与讨论
2.1 与自由基的反应机理
从表1可以看出,反应1~12两两互为竞争关系,为同种自由基在同一硅醇上的竞争反应。笔者先对这部分反应两两之间展开比较分析,判断优势反应,随后再与原甲烷氧化链式反应比较。
2.1.1 反应过渡态与能垒
反应1~11过渡态、虚频和反应能垒如图2所示。
以图2(e)中反应9,10为例,2者均为·OH在连生硅醇B位点上的反应,反应过渡态结构见上部结构图。经内禀反应坐标(IRC)验证,前者反应·OH从B位点抽取氢原子,生成H2O和连生硅醇自由基(缺H),反应过渡态虚频为1 503.638 7 cm-1;后者反应·OH从B位点抽取羟基,生成H2O2和连生硅醇自由基(缺OH),反应过渡态虚频为419.717 8 cm-1。反应能垒和能垒差见下部曲线图,反应10能垒更大(低温时将近8倍,高温时10倍左右),且其随温度升高速率明显大于反应9。其余反应同理。
反应12经反复尝试搜寻过渡态和柔性(刚性)扫描均无法发现合理反应路径,推测该反应不成立。由于受基团极性影响,·OH在该反应位点附近时,更容易受到B位点H原子吸引,而无法到达F位点合适反应位置。此外,连生硅醇分子间存在氢键作用,进一步加大了该反应的难度。综上,反应11无竞争反应,故图2(f)中只单独列出能垒分析。
此外,Ori_H和Ori_OH反应相关文献中已进行详细的分析[37],本文不再赘述,只在后续分析反应速率常数。
从能垒的角度分析,反应能垒越小,反应物越容易转变为过渡态物质,也越容易发生反应,但其并不是唯一决定反应快慢的量(如隧道效应也会影响反应速率)。严格意义上来讲,反应速率常数k才是从根本决定反应速率快慢的原因,下面对各反应速率常数进行分析。
2.1.2 反应速率常数
反应速率常数变化如图3所示。为方便比较各速率之间的影响,图中以lnk为y轴,以1 000/T为x轴。
在图3分析中,采用以Δlnk=2.3为影响界限(即范围在(0.1k,10k))。以Δlnk最小的反应5与反应6为例,2者最接近处位于1 273.15 K(最高温度),但劣势反应仍无法对优势反应产生明显影响。同理,图3中的反应1~10中,优势反应为反应1,3,5,7,9。其中反应1,5,7为硅醇与·H反应,反应3,9为硅醇与·OH反应(还应加上未参与比较的反应11)。
下面分别按照与·H或·OH反应的速率,比较上述反应与原链式反应Ori_H和Ori_OH速率常数大小。
由图4可知,2种硅醇与·H的直接反应速率均无法有效影响到原链式反应Ori_H。
当环境温度随甲烷氧化放热升温到31 ℃时(图4(b)中1 000/T=3.29 K-1处),反应9开始对链式反应产生影响,即开始减弱链式反应作用。随环境温度继续升高达到122 ℃时(图中1 000/T=2.53 K-1处),反应9的反应速率与Ori_OH相等,即两者争夺·OH的速率相等,链式反应得以有效减弱。环境温度进一步上升到259 ℃时(图中1 000/T=1.88 K-1处),反应9速率已为Ori_OH反应的10倍,链式反应得到有效抑制,且后续两者差距越来越大。此外,在温度为292 ℃时(图中1 000/T=1.77 K-1处),孤立硅醇与·OH反应达到与Ori_OH反应一样的速率。


图2 过渡态与能垒差Fig.2 Transition state and energy barrier difference

图3 反应速率常数Fig.3 Reaction rate constant
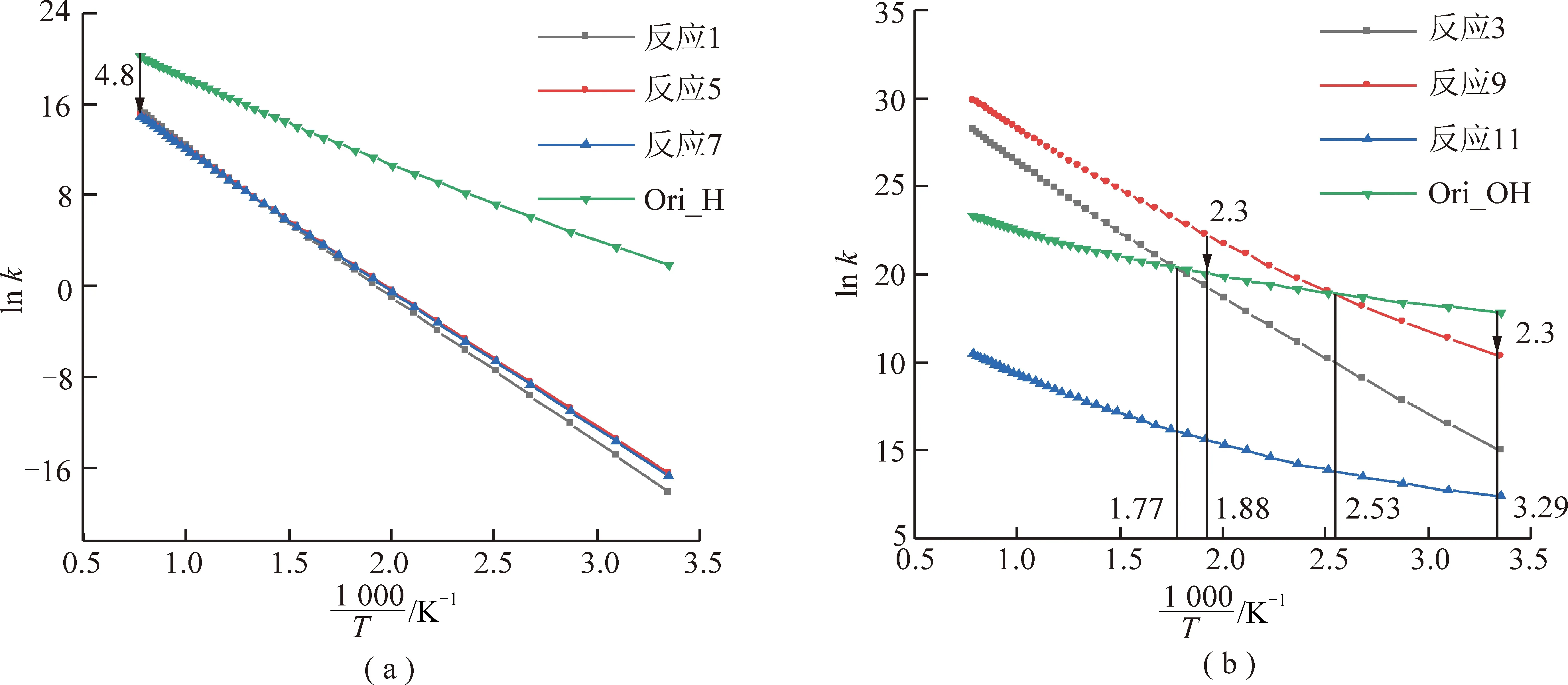
图4 反应速率常数比较Fig.4 Comparison of reaction rate constants
从前文可知,反应9的产物有2种:H2O和连生硅醇基(缺H)。由于连生硅醇基与·H恰好有合适反应位点,且自由基结合反应的高速特性,猜测Reverse反应成立,并对原链式反应有较强影响。通过过渡态搜寻和刚性扫描,确定该反应无过渡态。由于反应无过渡态,速率常数只能利用反应速率公式进行估算(以产物和反应物自由能之差代替能垒,透射系数取1)。估算结果表明:速率最大处为25 ℃时,数量级为10225;速率最小处为1 000 ℃时,数量级为1068;显然Reaverse反应速率远大于上述所有反应。另外,由于Reaverse反应依赖于反应9中连生硅醇基的产生速率,即可近似看作两反应速率相等,反应速率由反应9控制。二者反应相辅相成,反应原理如图5所示。

图5 反应流程Fig.5 Reaction flow chart
综上,硅藻土具有良好抑制甲烷氧化链式反应的效果,其中,连生硅醇在低温时便具备良好的性能。基于此,若能对天然硅藻土进行改性,增加其连生硅醇占比,将会大幅提高抑制效果。
2.2 对甲醛吸附作用
2.2.1 分子表面静电势(ESP)分析
从硅醇分子ESP(静电势)分布角度出发,硅醇与甲醛的可能吸附位点如图6所示。图中蓝色表示静电势为负,颜色越深数值越大;红色表示静电势为正,颜色深浅同理。此外,图中黄色小点表示硅醇分子静电势表面极大值点,青色小球表示极小值点。
由图6可以看出,孤立硅醇模型由于其规则的结构,仅在羟基侧方有与HCHO结合的位点(记为S)。针对连生硅醇模型,以羟基指向左方为正(图6(c)),分别初步确定了前(F)、后(B)、左(L)、右(R)以及上(U)5个位点。
2.2.2 吸附能计算与分析
各吸附位点构型和相应吸附能如图7所示。

图6 硅醇静电势极值点分布Fig.6 Distribution on extreme points of silanol electrostatic potential

图7 吸附构型及能量Fig.7 Adsorption configuration and energy
硅醇分子与HCHO吸附组成了HCHO-硅醇体系,使HCHO的分子运动减弱,降低了HCHO与自由基碰撞的概率,从而抑制后续链式反应的发生。
通过上表计算表明,连生硅醇的多个吸附位点中,U位点吸附能最大,且明显大于其他位点。连生硅醇相比孤立硅醇具有更强的吸附性能,更利于脱除链式反应中产生的HCHO。
2.2.3 吸附弱相互作用类型分析
利用Multiwfn软件进行弱相互作用可视化分析,结果如图8所示(为保证呈现效果,仅导出吸附位点局部图)。
由色带对比可知,孤立硅醇与HCHO的吸附作用由3种弱相互作用产生:氢键、范德华力和弱氢键。氢键发生在HCHO中的O原子与孤立硅醇H原子作用处,此处两分子间相互吸引最强;弱氢键发生在HCHO中H原子与孤立硅醇Si—O—Si处,吸引力中等;范德华力则夹杂在两者之间,由于距离各原子较远,吸引力最小。
连生硅醇与HCHO的吸附只有2种弱相互作用:氢键和范德华力。氢键发生在HCHO中的O原子与连生硅醇B位点H原子作用处,此处两分子间吸引力最大;范德华力发生在HCHO分子侧面相对连生硅醇分子处,吸引力较弱。通过对比原连生硅醇分子内的弱相互作用(图8(b),(c)),可知HCHO的加入加强了两羟基间的氢键作用,由发生吸附前的弱氢键作用转变为强氢键作用。

图8 弱相互作用可视化分析Fig.8 Visual analysis of weak interaction
综上,硅藻土对甲烷氧化链式反应中的关键中间产物HCHO具有较强的吸附效果。2种硅醇模型的吸附性能差别不大,均与HCHO结合形成氢键作用,其中双生硅醇吸附强度略大一点。
3 结 论
(1)针对甲烷氧化链式反应,硅藻土中的连生硅醇结构具备良好的抑制效果。在31 ℃时,便开始影响链式反应;在122 ℃时,达到相同速率,有效减弱链式反应;在259 ℃及更高温度时,有效抑制链式反应。
(2)硅藻土能与HCHO结合形成氢键体系,具备较好的脱除效果,其中连生硅醇在与HCHO的吸附,具有更稳定的吸附构型。
(3)不论是活泼的化学性质还是稳定的吸附构型,都离不开硅藻土表面的连生硅醇。若设计出经济高效的改性方法以提高硅藻土中连生硅醇占比,将会大幅增强抑制效果。
(4)参考硅藻土结构,以表面极性羟基更活跃为新型抑制剂的筛选或改性目标。
猜你喜欢
燃料化学学报(2023年3期)2023-03-11 03:34:40
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
电脑知识与技术(2018年3期)2018-03-21 09:27:04
资源节约与环保(2017年9期)2017-10-10 09:24:22
河北地质(2017年2期)2017-08-16 03:17:02
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
合成化学(2015年2期)2016-01-17 09:03:05
