
Sarm1基因在创伤性脑损伤中的作用
2022-11-11 02:03于欣宇车益军冉启山
遵义医科大学学报 2022年5期
刘 潜,于欣宇,车益军,曹 芳,冉启山
(1.遵义医科大学 研究生院,贵州 遵义 563099;2.遵义医科大学附属医院 神经外科,贵州 遵义 563099;3.遵义医科大学 贵州省普通高等学校脑科学特色重点实验室,贵州 遵义 563099)
创伤性脑损伤(Traumatic brain injury,TBI) 是神经外科常见的疾病之一。截止目前,TBI已经成为全球第三大致死、致残因素[1]。创伤性轴突损伤(Traumatic axonal injury,TAI)是TBI中最重要的亚型之一。研究表明,轴突断裂不是立即发生,而是一个迟发过程,在伤后4~24 h发生,这个过程被称为继发性轴突断裂,是轴突损伤的主要形式[2]。而在动物模型中发现,不管是经典的瞬间旋转损伤模型、自由落体损伤模型或控制性皮质损伤模型(Controlled cortical impact,CCI),都可引起不同程度的轴突损伤[1,3-4]。虽然随着CT、MRI、PET等临床影像诊断手段的进步,对TAI的诊断有一定的提高,但是免疫组化法检测脑损伤区β淀粉样前体蛋白(Amyloid precursor protein,APP) 聚集量,仍被认为是诊断TAI的金标准[3-4]。然而TAI的发生具体机制目前仍不清楚,目前仍缺乏特异而有效的药物及治疗手段。因此,研究TAI发生发展机制,抑制TBI后继发性轴突损伤、降解无疑是治疗TBI的重要策略,具有重要的临床价值及社会意义。
目前发现轴突损伤过程中有多种机制参与,其中Sarm1 (Sterile alpha and Toll/interleukin-1 receptor motif-containing 1) 在轴突变性中起到非常关键的作用,是轴突变性的主要执行者。最早由Osterloh等在果蝇(dSarm)及小鼠(Sarm1)轴突离断模型中发现,dSarm/Sarm1敲除后,可明显抑制轴突变性[5]。鉴于Sarm1在轴突变性中的关键作用,Sarm1在TAI中的作用逐渐受到关注[6]。TBI后敲除Sarm1可明显减轻TAI,但并未在活体小鼠验证Sarm1在TBI后是否通过抑制轴突损伤发挥神经保护作用[7]。因此,通过建立控制性皮质损伤模型(CCI),结合7T MRI及其他分子生物学技术,检测Sarm1在创伤性脑损伤尤其是创伤性轴突损伤中的作用,旨在为临床治疗颅脑创伤提供新的治疗方向。
1 材料与方法
1.1 试验动物、试剂及仪器 C57BL/6 雄性小鼠(Wild-type,WT,30只,10至12周龄,体重 22~25g),购买自长沙市天勤生物技术有限公司,许可证号为:SCXK(湘)2019-0014。Sarm1基因全敲除(Sarm1 knock out,Sarm1 KO)小鼠雄性、雌性各2只,共4只,来自于清华大学免疫学研究所,将上述4只Sarm1 KO小鼠作为种鼠,与C57小鼠杂交繁殖,繁殖后代经鉴定为纯合子用于本实验。Anti-Sarm抗体、Anti-PLK2抗体购自美国Abcam公司;颅脑创伤仪购自美国PSI公司;开颅手术器械均购自南京精工公司;7T MRI购自德国Bruker公司;倒置荧光显微镜购自日本奥林巴斯公司;转棒式疲劳仪ZB-200购自成都盟泰公司。
1.2 实验动物分组 实验用鼠共60只;C57BL/6 雄性小鼠30只,Sarm1基因敲除鼠(Sarm1 KO)30只。将两组小鼠分别随机分为野生型假手术对照组(WT-sham),野生型颅脑创伤组(WT-CCI),Sarm1基因敲除鼠假手术组(Sarm1 KO-sham),Sarm1基因敲除鼠颅脑创伤组(Sarm1 KO-CCI)。行为学评分使用鼠12只(6只WT,6只 Sarm1 KO),MRI扫描使用鼠12只(6只WT,6只 Sarm1 KO,与行为学评分使用鼠为同一批次小鼠)。行为学评分分别评价建模前与建模后1、3、5、7 d,MRI扫描于建模前与建模后1d分别进行。严格对照前期制定纳入标准,WB使用24只,免疫荧光和组化使用24只,行为学评分和MRI扫描使用12只,实验动物具体分组见表1。

表1 实验动物分组及动物数量(n=6)
1.3 小鼠TBI和假手术组模型的建立 使用3%戊巴比妥钠溶液作为麻醉剂,通过腹腔注射麻醉小鼠,头部清洁备皮,局部皮肤予以医用碘伏消毒;行头部矢状位右旁正中切口,剥离骨膜后暴露右侧颅骨术区,以前囟后2.0 mm,中线旁2.0 mm为钻孔位,用合适颅骨钻钻开3.0 mm的圆形骨窗,暴露顶叶。评估小鼠生命体征无异常后,将其头部固定在TBI-0310颅脑创伤仪上,选择2.0 mm直径的钝口打击头,予以打击,具体参数如下:打击速度为:5 m/s,接触时间为:100 ms,打击深度为:2.0 mm;造成实验小鼠右侧顶叶中重度脑挫裂伤,创伤完成后,予以术区彻底止血,缝合头皮,将小鼠放置在恒温箱中待麻醉复苏。假手术组除不接受颅脑损伤仪器打击外,其余操作和TBI小鼠完全一致。将上述4组小鼠建模后按照组别分笼放置饲养。
1.4 小鼠神经功能行为学评分(转棒疲劳实验) 运用双盲法对实验小鼠的运动功能评估。用小鼠转棒式疲劳仪(ZB-200,成都盟泰)检测小鼠的运动能力。建立CCI模型前夜让小鼠在转棒仪上练习1 min×3次,固定转速为16 r/min,间隔15 min。建模前1 h进行加速转棒实验,并详细记录小鼠转棒的基值。加速转棒实验从0 r/min开始,每隔10 s增加3 r/min,直到转速达到30 r/min。建模前及建模后第1、3、5、7天均进行加速转棒实验。并详细记录小鼠的落棒时间。若小鼠抱绕转棒2周无行走动作亦视为落棒。
1.5 Bruker 7T MRI扫描 借助7T小动物磁共振(Bruker bio spec USR 70/20,德国),选取TBI前1 d及TBI后第1天进行影像学检查。包括动物设置和序列优化,每只小鼠总成像时间为1.0 h。小鼠经气体麻醉后固定于MRI平台床上,体温控制在37.0 ℃,同时监测呼吸。分别在CCI损伤前行T2WI及弥散张量成像(Diffusion tensor imaging,DTI)扫描作为自身对照。损伤后24 h再次扫描。T2WI相比校损伤灶大小,各向异性分数(Fractional anisotropy,FA)评价轴突损伤程度。
1.6 冰冻切片和免疫荧光
1.6.1 冰冻切片 取材:于TBI造模后第1天,分别取WT sham组、Sarm1 KO-sham组、WT-CCI组和Sarm1 KO-CCI组小鼠各6只,深度麻醉,左心室内灌注4 ℃ PBS液置换体内血液直至肝脏和肠系膜血管完全变白后,继续予以4%多聚甲醛约50 mL进行灌注固定,确定小鼠完全死亡后,断头完整取出脑组织,分别置于4%多聚甲醛标本盒中,继续固定24 h。梯度脱水:固定完成的小鼠大脑依次分别置于10%、20%、30%浓度的蔗糖溶液中,4 ℃环境下分别脱水24 h。切片制备:脱水完成的完整脑组织沿冠状位切片,直至创伤灶区域胼胝体,修正整齐,放入包埋盒,加入OCT包埋剂,排除气泡,超低温迅速冷冻,然后将其固定于冰冻切片机,调整好方向,15 μm厚度,冠状位连续切片,将切片展开,依次贴附在防脱载玻片上,做好标记,常温晾干,-20 ℃冰箱保存备用。
1.6.2 免疫荧光 复温:取冻存的冰冻切片放置于湿盒内,在常温下复温30 min。清洗:将制备复温后的切片浸入装满PBS的洗片盒中,再放置于水平摇床上(参数为:45 r/min,10 min),重复3次。破膜:将浓度为0.03%曲拉通,滴注于切片上,使其完全覆盖整张脑组织切片,置于湿盒中20 min,然后重复上述清洗步骤。抗原封闭:将抗原封闭液轻柔的滴注在切片上,常温放置于湿盒1 h,封闭完成。孵育一抗:用滤纸小心吸除切片上附着的封闭液,滴入稀释好的一抗,置于湿盒中,4 ℃冰箱中静置保存过夜,重复上述清洗步骤。孵育二抗:取出已完成孵育一抗切片,置于室温中复温20 min,然后进行梯度脱水,接着滴加二抗,常温下置于湿盒避光孵育2 h,完成后再次予以清洗。染核:脑组织切片滴加DAPI,浸染10 min,然后清洗。封片:滴加抗荧光淬灭剂,盖玻片轻推载玻片,避免气泡产生。观察:将制备好的切片置于荧光显微镜下观察,拍照。
1.7 免疫印迹法(Western blot) 配胶: 使用SDS-PAGE凝胶快速配制试验试剂,其中下层分离胶浓度12%,上层胶浓度为5%。上样:将制备好的蛋白样品加入上样缓冲液100 ℃沸水煮浴5 min,待冷却后移液器上样。电泳:将电压调整至80 V,电泳至蛋白分离后,调整电压为120 V,总时长约1 h。转膜:切取对应分子量大小的凝胶,放置于浸有预冷电转液的滤纸上,覆盖上相应大小的PVDF膜置于冰水浴中,恒定电流200 mA电转1 h,将蛋白转移至FVDP膜上。封闭:取出转膜完成的PVDF膜,放入5%的脱脂牛奶中进行封闭,常温下置于水平摇床(参数:45 r/min,1 h)。孵育一抗:取封闭好的PVDF膜快速放入盛有按比例稀释好的一抗抗体孵育盒中,置于4 ℃恒温冰箱水平摇床过夜。孵育二抗:取出PVDF膜,用TBST缓冲液,常温下水平摇床90 r/min清洗4次,常温下孵育2 h。显影:将孵育完成后的PVDF膜放置于凝胶化学发光成像系统,加入适量配制好的ECL显影液,曝光得到免疫印迹带,用于后续分析统计。
1.8 免疫组化染色 复温切片:将制备完成的脑组织切片,厚度为 5~6 μm,室温放置30 min后,放入4 ℃丙酮固定10 min,然后用PBS反复清洗,每次5 min,共3次,后用3%过氧化氢避光孵育20 min,消除内源性过氧化物酶的活性,再次用PBS清洗2次,每次5 min。正常血清封闭:从染片缸中取出切片,擦净切片背面水分及切片正面组织周围的水分,保持组织呈湿润状态,滴加正常兔血清处理,置于37 ℃环境下15 min。滴加一抗:用滤纸吸去血清,不洗,直接滴加一抗,置于4 ℃冰箱过夜。取出切片用PBS清洗2次,每次5 min (置于摇床)。滴加二抗:滴加生物素化的二抗,置于37 ℃环境下40 min,后再用PBS清洗2次,每次5 min (置于摇床) 。DAB显色:镜下观察,适时终止,用自来水充分冲洗;苏木素复染,室温,30 s,自来水充分冲洗。梯度酒精脱水:80% 2 min,95% 2 min,100% 2次,5 min。二甲苯透明:I,II (二甲苯)各5 min封片,最后用加拿大树胶(或中性树胶)封片。
1.9 统计学处理 运用SPSS 18.0软件对数据进行统计分析,转棒实验采用Two-way ANOVA分析,余检测采用One-wayANOVA分析,P<0.05认为差异具有统计学意义;采用GraphPad prism 7.0软件制作图表。
2 结果
2.1 小鼠转棒实验 为了解WT组和Sarm1 KO组小鼠造模前后神经运动功能情况,选择转棒实验进行验证,结果显示(见图1),在造模前,与WT组比较,两组行为学无明显差异(P>0.05,n=6),表明Sarm1基因敲除后不影响小鼠运动功能。CCI损伤后小鼠转棒时间较造模前明显降低,同时,与WT组比较,Sarm1 KO组小鼠在伤后第1天(P<0.01,n=6),第3天(P<0.01,n=6),第5天(P<0.05,n=6)小鼠运动功能显著改善。同时可以发现,在创伤后第1天,小鼠神经运动功能损伤最重,故选择伤后第1天(24 h)进行后续实验。
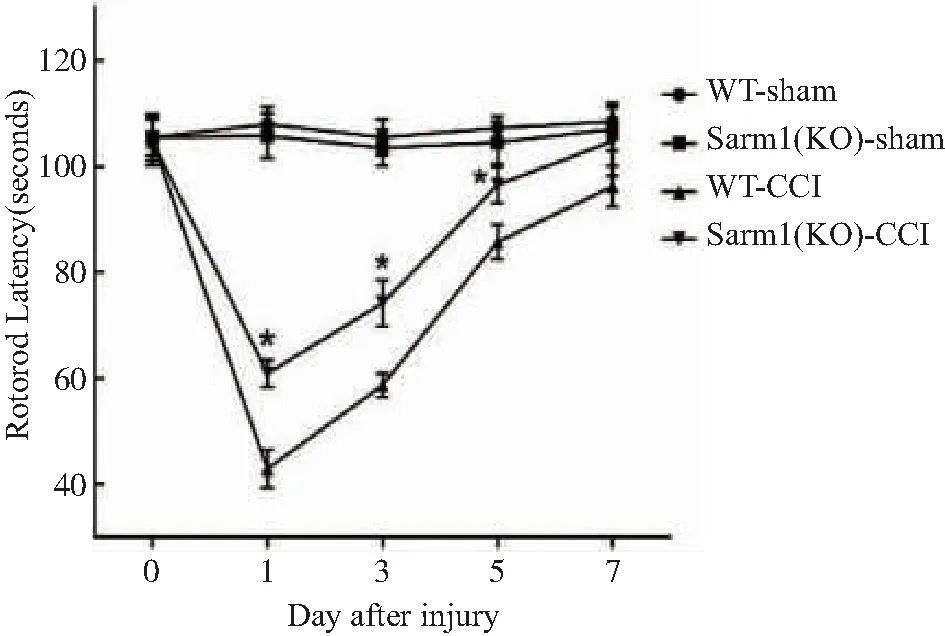
*:WT-CCI组与Sarm1 KO-CCI组比较,P<0.05。图1 创伤前、后1、3、5、7 d小鼠转棒时间
2.2 Bruker 7T MRI 扫描
2.2.1 CCI后Sarm1敲除对损伤灶大小的影响 7T MRI评价野生型小鼠与Sarm1基因敲除小鼠在脑损伤前后创伤灶范围大小差异情况。选择创伤前1d和创伤后第1天进行对比(见图2)。相较于对照组,实验组创伤后第1天(WT-CCI、Sarm1 KO-CCI)术区皮层脑组织均出现不同程度组织创伤;比较两组损伤灶大小,Sarm1 KO-CCI组小鼠损伤灶较WT-CCI组明显减小,差异有统计学意义(P<0.01,n=6)。
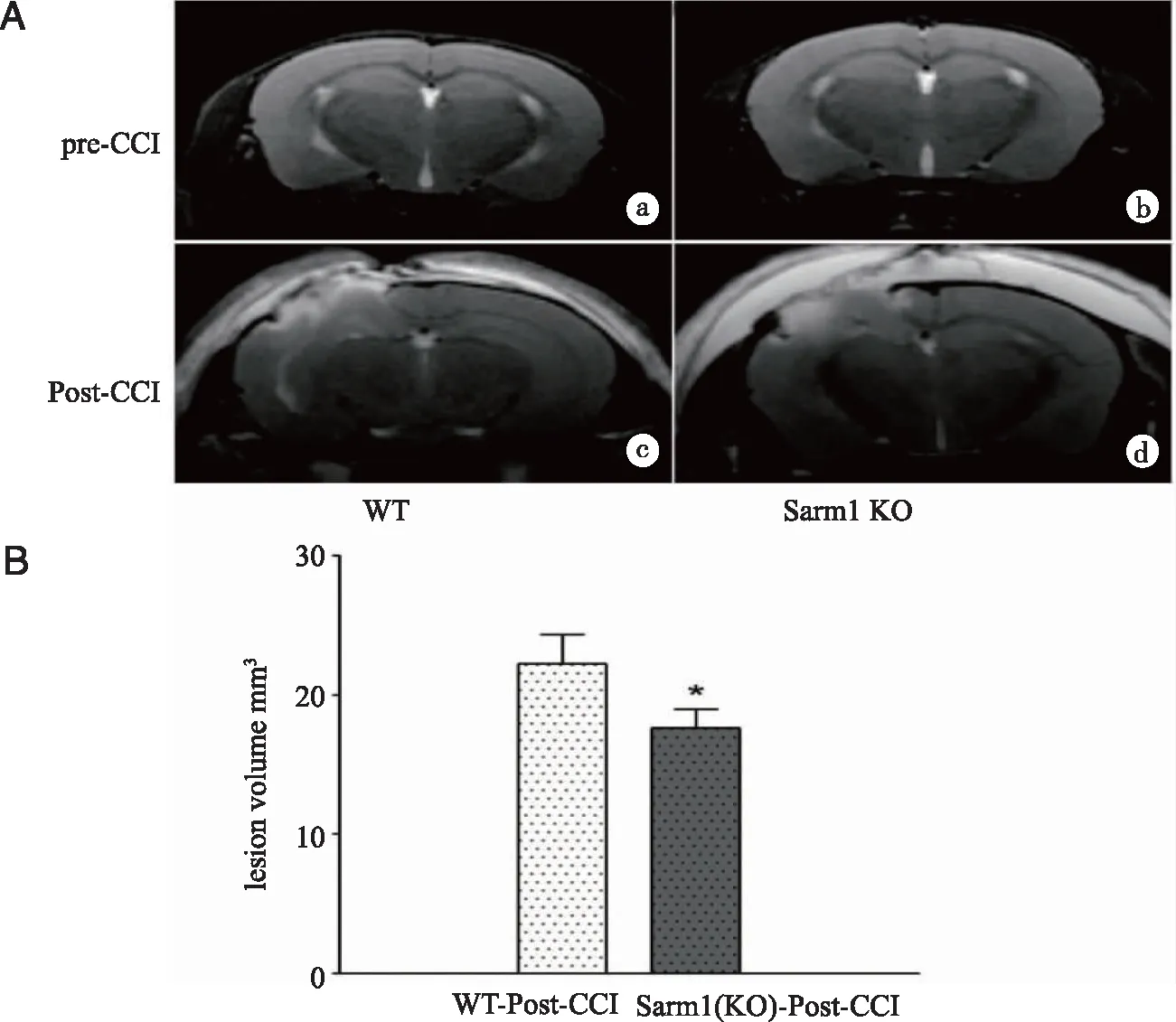
A:a:WT-sham组T2图像;b:Sarm1 KO组T2图像;c:创伤后1天(WT Post-CCI组)T2图像;d:创伤后1天(Sarm1 KO-Post-CCI)组T2图像; B:*:WT-CCI组和Sarm1KO-CCI组比较,P<0.01。图2 两组脑创伤前、创伤后第1天MRI T2图像
2.2.2 CCI后Sarm1敲除对轴突损伤的影响 为了研究WT型小鼠与Sarm1 KO小鼠在脑损伤前后轴突损伤的表达变化差异,分别对实验小鼠建模前1d和建模后第1天进行7T MRI DTI扫描,选择伤侧部位胼胝体区为感兴趣区检测FA值(见图3)。正常状态下,相较与WT组,Sarm1基因敲除后不影响FA值,说明敲除Sarm1不造成轴突损伤。CCI后,两组小鼠FA值明显降低,但Sarm1 KO组小鼠FA值降低明显受到抑制(P<0.05,n=6),表明Sarm1敲除后明显抑制抽突损伤。
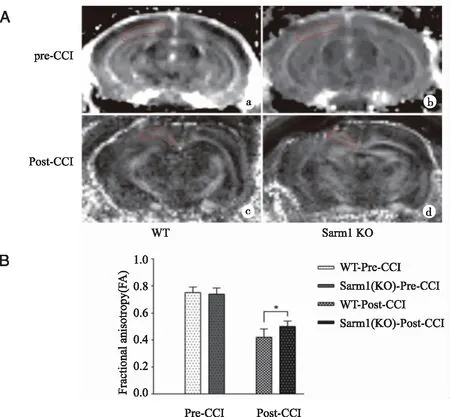
A:a:创伤前1天WT组DTI图像;b:创伤前1天Sarm1 KO组DTI图像;c:创伤后1天WT组DTI图像;d:创伤后1天Sarm1 KO组DTI图像;B: *: WT -CCI组与Sarm1KO-CCI组比较,P<0.05。图3 两组小鼠脑创伤前后1天的DTI图像
2.3 不同方法验证APP的表达
2.3.1 免疫荧光法验证APP的表达 病理学检查APP表达水平是检测是轴突损伤的金标准,故而我们对CCI后的创伤部位的APP的表达水平进行检测。在脑创伤前,WT-Sham组和Sarm1 KO-Sham组中,轴突损伤特异性标记因子APP未见明显表达,表明敲除Sarm1不造成轴突损伤;CCI后,两组小鼠APP表达均明显增高, Sarm1 KO-CCI组较WT-CCI组APP表达明显减少,表明敲除Sarm1后,明显抑制APP过表达(P<0.05,n=6,见图4)。
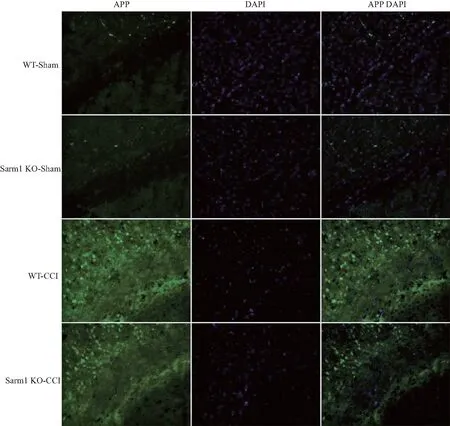
绿色荧光标记APP、蓝色荧光标记细胞核(DAPI染色,标尺=500 μm)。图4 两组在创伤前、后1 d APP在胼胝体区的表达水平
2.3.2 Western blot验证APP的表达 APP在正常WT组及Sarm1 KO组小鼠脑组织的表达量均较低,两组间无明显差异,表明Sarm1敲除后不造成明显的轴突损伤。CCI后第1天,两组APP表达相较于对照组均有明显升高(P<0.01,n=6),且Sarm1 KO-CCI组较WT-CCI组表达明显降低(P<0.05,n=6,见图5),表明Sarm1敲除后明显抑制APP过表达,抑制轴突损伤。
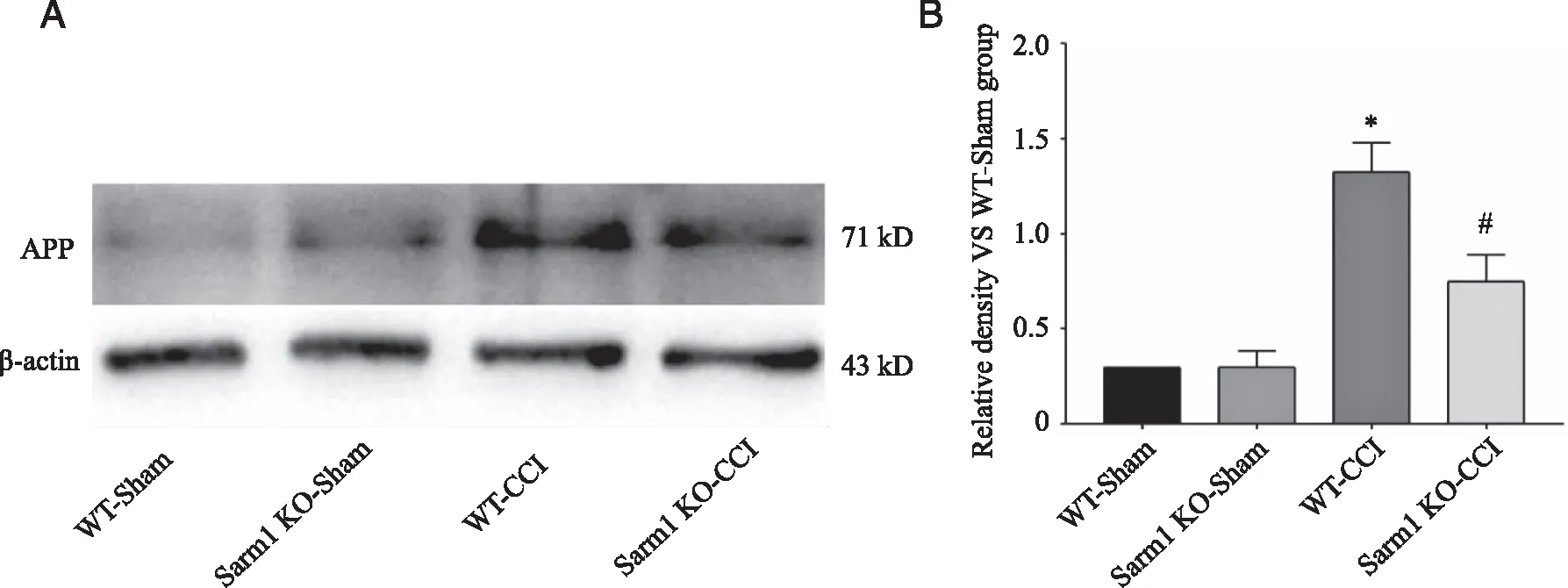
A:蛋白条带;B:统计分析结果;*: WT-CCI组与WT-sham组比较,P<0.01;#: Sarm1 KO-CCI组与WT-CCI组比较,P<0.05。图5 Sarm1敲除CCI后APP的表达
2.4 NeuN染色检测Sarm1对CCI后神经元活性的影响 取CCI损伤灶靠中线周边皮质区为感兴趣区,对照组取对应皮质区,每只小鼠选取 3 张切片,每张切片选取互不重复的 3 个视野(×200) ,采用Image-Pro Plus 6.0 软件计数NeuN阳性神经元数(见图6A)。结果显示,正常组中,Sarm1敲除后不影响NeuN表达。CCI后第1天,两组小鼠NeuN表达均明显降低(P<0.05,n=6),且Sarm1 KO组较WT组损伤灶周边NeuN表达明显增高(P<0.05,n=6,见图6B),表明敲除Sarm1后明显抑制神经元死亡。
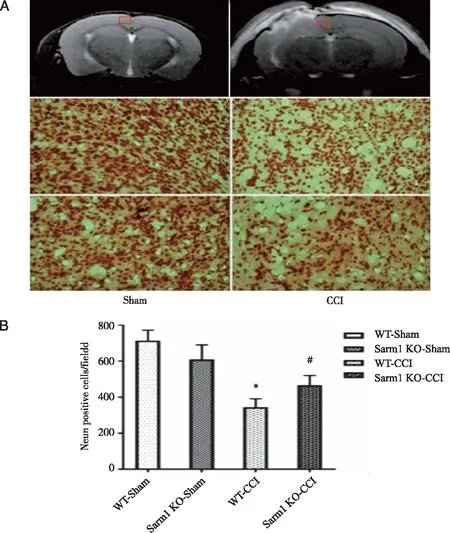
A:MRI T2,红色标记内为NeuN染色取材部位;B:统计分析结果;* :WT-CCI组与WT-sham组比较,P<0.05;#:Sarm1 KO-C CI组与WT-CCI组比较,P<0.05。图6 Sarm1对CCI后神经元活性的影响
3 讨论
APP是一种跨膜糖蛋白,是神经细胞的正常组成部分。它由高尔基体处理,并沿轴突快速向前运输,其功能包括细胞粘附、生长、神经保护和对损伤的反应,能促进轴突萌发、突触发生、神经突生长[8]。大量文献证实,APP是目前检测轴突损伤的金标准[9],其在伤性脑损伤后不到2 h就可识别出阳性反应,可作为TAI后早期检测指标[10]。本研究通过免疫荧光及WB实验检测证实,Sarm1可抑制CCI后APP过表达,表明Sarm1明显抑制TBI后轴突损伤。
鉴于DTI对大脑微结构改变高度敏感,是活体检测实验性动物和人类TBI后常用的影像生物标志物[11]。我们检测了胼胝体 DTI 指标FA值的变化,对损伤前后24 h的创伤小鼠观察,发现损伤前两组间FA值无明显差异,表明Sarm1敲除后明显抑制轴突损伤;而在建模成功后第1天,两组FA值均明显降低,Sarm1敲除后明显抑制FA值降低,表明敲除Sarm1明显抑制轴突损伤。
NeuN是一种在多数中枢和周围神经系统的有丝分裂后神经元中表达的胞核蛋白[12],在脑椎体神经元和颗粒性神经元表达,能特异性与神经元细胞核的抗原结合,是一种良好的成熟神经元标记物[13]。这种神经元蛋白最初是使用一种称为NeuN的单克隆抗体通过免疫细胞化学反应被鉴定。通过分析,NeuN被鉴定为Fox-3基因产物[14]。Fox-3包含一个RNA识别基序,并作为一种剪切调节分子发挥作用[14-15]。Fox-3调节NumB的选择性剪切,进而促进发育过程中的神经元分化。研究发现,TBI后损伤灶周边NeuN表达量增加与神经功能改善成正相关[16-17]。本研究证实,正常状态下,Sarm1敲除后并不影响NeuN表达,而CCI后,NeuN表达明显降低,Sarm1敲后明显抑制NeuN表达降低,表明Sarm1敲除后明显抑制CCI后神经元死亡。
正常情况下,Sarm1在神经元的形态学上发挥截然不同的作用。虽然发育时,Sarm1参与神经元的形态发生,但是Sarm1敲除鼠可以正常发育,表现出正常的运动功能[18],这与我们的研究结果一致。而在离体神经元损伤模型中,无论是线粒体毒性损伤还是氧糖剥夺,都可以高表达及激活内源性Sarm1从而促进神经元死亡及轴突变性[19]。而外源性Sarm1,可促进神经元细胞死亡[20]。缺氧后Sarm1可以促进运动神经元变性死亡[21],而这种死亡不同于凋亡、坏死,被称为Sarm1依赖性死亡[19]。近期研究证实,Sarm1是一种内源性NAD+分解酶,活化的Sarm1可直接将NAD+分解为ADP-ribose (ADPR),cyclic ADPR和烟酰胺(Nicotinamide)[22]。Sarm1通过分解NAD+在能能量代谢中起到了关键作用,在多种神经系统急慢性模型中(比如在阿尔茨海默病、缺血性脑卒中以及创伤性脑损伤)促进轴突损伤及神经元死亡[22-25]。本研究通过活体小鼠MRI扫描及其他分子生物学方法证实,CCI后,Sarm1敲除后通过抑制轴突损伤与神经元死亡,发挥神经保护作用。
综上所述,在创伤性脑损伤后,敲除Sarm1可减轻脑组织损伤,抑制轴突损伤及神经元死亡从而发挥神经保护作用。
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
浙江临床医学(2021年12期)2021-11-29
江西医药(2020年4期)2020-04-28
幸福(2019年21期)2019-08-20
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
标记免疫分析与临床(2016年9期)2016-11-21
中国康复(2015年4期)2015-04-10
郑州大学学报(医学版)(2015年2期)2015-02-27
中华神经创伤外科电子杂志(2015年1期)2015-01-21
