
脒类化合物在构筑含氮杂环骨架中的应用进展
2022-11-06 11:43孙振威庹文君贾丰成
武汉工程大学学报 2022年5期
孙振威,罗 娜,庹文君,张 翔,贾丰成*,2
1.武汉工程大学化学与环境工程学院,湖北 武汉 430205;
2.绿色化工过程教育部重点实验室(武汉工程大学),湖北 武汉 430205
脒类化合物是一种廉价易得的重要有机合成砌块,广泛地用于各种氮杂环的合成。一般来说,脒既可以作为双原子合成砌块,也可以作为三原子的合成砌块。由于其具有两个氮亲核位点,脒广泛地作为双亲核试剂参与反应,同时由于内部特有的亚胺单元,也可以充当亲核亲电试剂使用。正是因为脒类化合物丰富的合成功能,使之成为了制备多种氮杂环如吡咯、嘧啶、三嗪、咪唑、吲哚、苯并咪唑、喹啉、喹唑啉和噻唑类杂环化合物的关键前体。近10 年来,伴随有机合成领域新试剂、新策略以及产生活性中间体的方式发生了巨大的变化,来源广泛的经典底物相关的合成方法学迎来了飞速的发展浪潮。脒盐作为一种廉价易得的大宗原料,与此相关的合成反应也取得了阶段性长足的进展。本文着重综述了近10 年来脒类化合物参与的串联环化反应,主要包括无过渡金属反应体系以及自由基型环化反应,并对相应的反应策略和机理进行详细阐述。
1 无过渡金属体系
2015 年,邓国军等报道了一种无过渡金属条件下,以脒盐和酮类化合物为原料高效生成4,5-二氢-1H-咪唑-5 酮的串联重排反应[1-6]。机理研究表明,首先,酮与脒缩合生成中间体1A,中间体1A烯胺α位在碱的作用下脱质子形成碳负离子,对氧气加成生成超氧阴离子1B,接着超氧阴离子脱去一个氢氧根离子,形成酰亚胺中间体1C。在碱的作用下,1C 失去质子生成亚胺离子1D。接下来亚胺离子直接对羰基的碳原子进行亲核加成形成中间体1E。1E 通过逆adol 反应得到中间体1F。最后从水中捕获一个质子得到目标产物3a(图1)。

图1 碱促进脒盐和酮参与的串联环化反应Fig.1 Base-promoted tandem cyclization reactions of amidine salts with ketones
2015 年,Chaskar 课题组报道了一种绿色串联环化-氧化合成多取代嘧啶骨架的方法[7-9]。反应以α,β-不饱和酮为原料,以循环利用的氢氧化胆碱(ChOH)为催化剂,在加热条件下直接和脒盐酸盐进行Michael 共轭加成形成中间产物烯醇2A,生成的烯醇通过氢氧化胆碱(ChOH)氢键稳定。2A 质子化再互变异构产生亚胺中间体2C,其亚胺部分对羰基分子内亲核加成产生2D,2D 互变异构得到中间体2E。然后,在碱的作用下2E 脱水产生二氢嘧啶2F。最后,在氧气作用下进一步氧化芳构化形成最终产物多取代嘧啶6a(图2)。
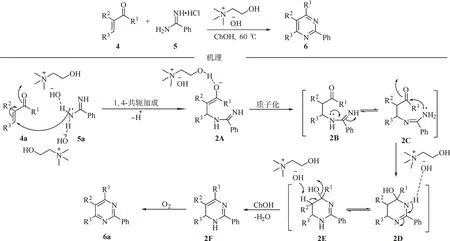
图2 氢氧化胆碱催化α,β-不饱和酮与脒盐的串联环化反应Fig.2 ChOH-catalyzed tandem cyclization of α,β-unsaturated ketones with amidine salts
2016 年,吴安心等率先发展了一种在TBHP/K3PO3协同促进的氧化环化反应,用于构建喹唑啉酮衍生物[10-11]。该方法采用靛红和脒盐为反应底物,在室温条件下以良好至中等的产率得到喹唑啉酮衍生物。通过一系列控制实验表明,靛红在TBHP 的作用下发生Baeyer-Villiger 氧化生成重要的靛红酸酐中间体3C。继而脒盐酸盐在碱的作用下,对靛红酸酐亲核进攻脱除CO2生成酰胺3D。最后通过脱氨环化反应得到目标产物8a。作者发现在不使用脒盐的条件下,靛红可以发生类似的自二聚反应得到天然产物色胺酮及其衍生物(图3)。

图3 过氧叔丁醇介导靛红和脒盐参与的氧化扩环反应Fig.3 TBHP-mediated oxidative ring expansion reactions of isatins with amidine salts
2018 年,吴安心等开发了一种高效的K2S2O8介导的四组分氧化环化反应,用于合成嘧啶衍生物[12]。该反应以甲基酮、脒盐和DMSO 为底物,通过一锅法可以形成2 个C-C 键和2 个C-N 键。该反应的特点是DMSO 作为双合成子参与环化反应,即甲基和次甲基单元前体。作者结合控制实验提出了以下机理路径:最初,DMSO 被K2S2O8激活生成原位硫正离子4B,该硫正离子立即被烯醇捕获,生成3-(甲基硫)-1-(对甲苯)丙-1-酮。消除MeSH 生成α,β不饱和酮。随后,盐酸苄脒分子间氮杂-迈克尔加成得到中间体,分子间缩合转化为环状中间体。分子内消除甲基硫醇和双键的互变异构化,得到所需的产物。值得注意的是,作者并未检测到可能的中间体7,但鉴于早期文献中甲基酮可以与DMSO 偶联产生β-甲硫基异丙烯基酮,据此作者还提出了另一可行途径。中间体4D 与亲核有机碱DABCO 发生1,4 加成形成中间体4G。4G 对硫正离子亲核加成和发生三级胺的消除形成中间体4H。然后4H 与4a 发生环化反应得到中间产物4I,4I 经脱甲硫醇和互变异构化反应最终得到预期的产物11a(图4)。

图4 过硫酸钾介导甲基酮、二甲基亚砜和脒盐参与的四组分氧化环化反应Fig.4 K2S2O8-mediates four-component oxidative cyclization reaction of methyl ketones,DMSO and amidine salts
2019 年,江焕峰等使用三氟甲基酮和脒盐为底物,报道了一种在空气中选择性C-C 键断裂合成5-三氟甲基化咪唑-4-酮的新方法[13-15]。通过控制实验提出了可能的反应机理:首先,脒与三氟甲基酮在碱的作用下发生缩合生成中间体5A,中间体5A 在亚胺型和烯胺型之间可以发生平衡互变。随后,在碱和氧气的协同作用下,亚胺中间体5A在碱性条件下与氧气结合生成超氧阴离子5B,接着过氧键断裂脱去一个氢氧根离子后转化为羰基中间体5C。氮原子分子内进攻其羰基位置得到中间体5D,再经过1,2-芳基迁移生成中间体5E。最后,中间体5E 经异构化和质子化反应生成目标产物14a(图5)。

图5 包含碳-碳键切断的三氟甲基酮和脒参与的串联环化反应Fig.5 Tandem cyclization reaction containing C-C bonds cleavage of trifluoromethyl ketones with amidine salts
2019 年,王存德课题组发展了DBU 介导的2-酰基-1-氰环丙烷羧酸酯与脒盐的串联环化反应,合成多取代嘧啶衍生物[16]。基于该课题组前期工作,提出了一种可能的反应路径。首先,脒盐酸盐在DBU 介导下形成游离脒,与底物2-酰基-1-氰环丙烷羧酸酯的羰基位发生缩合反应生成亚胺中间体6A。由于其亚胺作为缺电子单元从而促进中间体6A 的C-C 键选择性断裂,导致了碳鎓离子中间体6B 的生成。由于中间体6B 不稳定,乙氧羰基容易以二氧化碳和氯乙烷的形式消除,得到共轭氰基丁二烯中间体6C。随后6C 夺去DBUH+的质子形成具有稳定共轭体系的亚胺盐离子6D,中间体6D 通过分子间迈克尔加成环化生成氰基二氰嘧啶中间体6E,最终6E 消除氰化氢生成所需产物17a(图6)。

图6 1,8-二氮杂二环十一碳-7-烯介导的2-酰基-1-氰环丙烷羧酸酯与脒参与的串联环化反应Fig.6 DBU-mediated tandem cyclization of 2-acyl-1-cyanocyclopropane carboxylates with amidine salts
2019 年,宋秋玲课题组采用溴二氟乙酸乙酯作为二氟卡宾前体,可以在无过渡金属条件下发生四重裂解产生二氟卡宾与脒盐反应,为具有重要药物价值的1,3,5-三嗪类化合物的构建提供了一种新途径[17]。通过二氟卡宾捕获实验,他们提出了一条合理的反应机制。首先,在碱的作用下,溴二氟乙酸乙酯经皂化反应水解成其羧酸盐后,然后在加热条件下脱羧和脱溴原位生成二氟卡宾。随后,二氟卡宾被脒捕获得到中间体7A,在碱性条件下通过一次脱氟反应转化为亚胺中间体7B。紧接着,活性中间体7B 受到另一个脒分子的亲核加成,发生第二次脱HF 后生成共轭亚胺中间体7C。最后中间体7C 经历分子内亲核加成,通过消除一个氨分子产生目标产物20a(图7)。

图7 溴二氟乙酸乙酯充当二氟卡宾前体与脒盐的串联环化反应Fig.7 Tandem cyclization of ethyl bromodifluoroacetates acting as difluorocarbine precursor with amidine salts
2021 年,江桃山课题组开发了一种通过以底物脒盐作为DMSO 活化剂,为嘧啶衍生物的合成提供一种新方法[18]。具体的机理为DMSO 被脒盐酸盐活化,原位生成硫正离子8A,随后被烯胺8B 亲核进攻得到化合物8C。化合物8C 容易发生消除甲硫醇得到α-亚甲基化产物8D。最后中间体8D 与脒盐酸盐发生迈克尔加成,分子内缩合。最后在空气作用下氧化芳构化得到目标产物23a(图8)。

图8 融合底物脒活化二甲基亚砜步骤的串联环化反应Fig.8 Tandem cyclization reaction involving DMSO activated by amidine salts
2020 年,高庆贺等报道了一例以DTBP 和分子氧为氧化剂,I2催化的醛、脒和三级胺参与的多组分串联环化反应[19]。该反应通过DTBP/I2氧化三级胺生成烯胺中间体,将叔烷基胺的α-官能化转化为α,β-双官能化,为构建嘧啶骨架提供了一种新颖有效的途径。在控制实验基础上,作者提出了一种可能的机理。首先,26a 在I2和DTBP 的共同氧化作用下生成关键亚胺中间体9A,通过互变异构化原位生成烯胺中间体9B。醛可以捕获烯胺中间体9B,从而避免亚胺中间体9A 与脒反应。随后,将24a 亲核加成到具有更高亲电性的α,β-不饱和亚胺9C 得到中间体9D。最后,中间体9E 经过分子内脱胺、环化和氧化芳构化反应得到目标产物27a(图9)。

图9 碘催化醛、脒和三级胺参与的多组分环化反应Fig.9 I2-catalytic multicomponent cyclization reactions of aldehydes,amidine salts and tertiary amines
2022 年,沈志良课题组研究了无过渡金属条件下碱促进脒盐和全氟烷基炔烃的[3+3]环加成反应,为全氟烷基取代的嘧啶衍生物合成提供一种新方法[20-21]。在一锅法中,脒经过氢胺化/脱氟/环化过程,构建2 个新的C-N 键和一个新的杂环。该反应的可能反应机理:首先,在碱的作用下将脒选择性加成到全氟烷基炔的三键上生成乙烯基阴离子10A,其从水中攫取1 个质子生成氢胺化中间体10B。随后,分子内亚胺在碱的作用下拔氢进行分子内亲核进攻,导致C-F 键裂解得到环状分子10C。10C 在碱的辅助下,再消除1 个HF 分子生成全氟烷基嘧啶衍生物30a(图10)。

图10 碱促进脒盐和全氟烷基炔烃的[3+3]环加成反应Fig.10 Base promoted[3+3]cycloaddition reactions of amidine salts with perfluoroalkynyls
2 自由基型环化反应
2015 年,江焕峰课题组开发了一种铜催化的脒盐和甲基芳烃的级联环化反应,为3,5-二取代-1,2,4-噁二唑的合成提供一种新策略[22]。该反应以铜作为催化剂,TBHP 作为氧化剂,磷酸钾作为碱,室温下反应12 h,即可构建1,2,4-噁二唑类衍生物。此反应具有原子经济、操作简单、官能团耐受性好的优点。在控制实验和以往文献的基础上,该反应的可能机理路径是,首先,甲苯被Cu/TBHP 体系作用下形成酰基自由基11A,并与脒发生自由基加成生成中间体11B。随后,二价铜夺去中间体的1 个单电子变成一价铜后,自由基中间体11B 发生单电子氧化生成碳正离子中间体11C。然后,失去1 个质子得到酰胺中间产物11D,经过烯醇异构化转化为中间体11E。中间体烯醇结构与铜催化剂配位得到配位化合物11F。最后,随着H+释放,NH 与铜中心结合生成中间体11G,经过还原消除得到目标产物23a(图11)。

图11 铜催化脒和甲基芳烃参与的串联环化反应Fig.11 Copper-catalyzed tandem cyclization reactions of amidine salts with methyl aromatics
2016 年,邓国军等报道了由脒类、单质硫、2-甲基喹啉参与的三组分反应[23-26]。在无过渡金属条件下,以磷酸钾为碱,120 ℃下以中等到良好的收率合成一系列1,2,4-噻二唑衍生物,且具有较好的区域选择性,强的官能团耐受性。该反应的机理是:首先,硫氧化2-甲基喹啉生成碳自由基中间体12A,对脒进行自由基加成,然后与单质硫发生单电子转移,失去一个质子形成中间产物12B。接下来的步骤是,中间产物上的胺对S8环亲核进攻,生成多硫化胺中间体12C。然后中间体亚胺异构化反应得到中间体12D,中间体12D 的强亲核末端硫引发分子内脱硫,致使与N 相连得S位点亲核进攻亚胺得到4H-噻二唑12E。最后,被当量硫氧化得到目标产物26a(图12)。

图12 脒类、单质硫和2-甲基喹啉参与的自由基型串联环化反应Fig.12 Radical-type tandem cyclization reactions of amidines salts,monosulfur and 2-methylquinolines
2016 年,江焕峰等报道了烯丙基C(sp3)-H 和乙烯基C(sp2)-H 在碱的促进作用下与脒盐发生双重C-N 键成键的反应,从而构筑多种多取代嘧啶化合物[27-30]。反应表现出了良好的官能团耐受性,而且该策略无需使用过渡金属,以氧气作为唯一氧化剂。基于控制实验和前期工作,提出一种可能的反应路径,苯甲脒在碱的作用下脱质子形成中间体13A,随后在氧气的作用下经过单电子转移过程,得到超氧阴离子自由基和自由基中间体13B。然后,芳基烯丙基通过氧化生成高度稳定的烯丙基自由基13C,随之与中间体13B 交叉偶联得到中间体13D。最后,13D 经历去质子化-氧化-加成和芳构化过程形成产物29a(图13)。
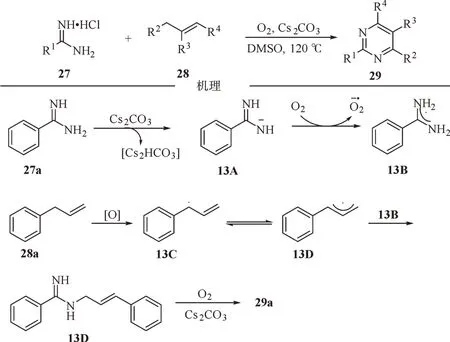
图13 碱促进下烯丙基化合物与脒的氧化环化反应Fig.13 Base-promoted oxidative cyclization of allyl compounds with amidine salts
2017 年,纪顺俊/徐小平课题组报道了一种铜催化吲哚和脒盐的分子间氧化环化反应,为八元氮杂环骨架的高效构筑提供了一种新途径[31]。他们采用氧气作为氧源,NMP 为溶剂,80 ℃的条件下能以中等到优秀的收率得到苯并[1,3,5]三唑嗪-6(5H)-1 衍生物。可能的反应路径是,首先,2-芳基吲哚在氧气作用下掘氢经历自由基路径生成吲哚自由基中间体14A。然后,在分子氧的存在下,形成位于C3 的吲哚过氧化物14C。之后从吲哚中掘氢脱水得到中间体吲哚酮14E。随后,中间体14E 与脒盐可能经历两种不同路径得到反应产物。一种是苯甲脒对吲哚酮14E 进行亲核进攻,形成中间体14F,脒上的胺对吲哚酮上的羰基再进行一次分子内亲核进攻。最后,发生分子内电子转移,C-C 键断裂开环得到目标产物32a。另一种途径是,中间体14E 经过Baeyer-Villiger 氧化过程得到中间产物14I。随后,苄脒对羰基进行亲核进攻,继而发生C-O 键断裂开环,得到酰胺中间体14K。最后,分子内的关环通过分子内脱水缩合反应生成目标产物32a。2019 年,该课题组以Eosin Y-Na2为催化剂,实现了可见光诱导下2-芳基吲哚和脒盐参与的扩环反应,同样可以高效构建苯并八元氮杂环类化合物[32]。作者也证明了机理与上述铜催化体系的扩环反应相类似(图14)。

图14 铜催化吲哚和脒盐参与的氧化扩环反应Fig.14 Copper-catalyzed oxidative ring expansion reactions of indoles with amidine salts
2017 年,江焕峰等报道了一例铜催化的脒盐和丙酮类衍生物参与的[3+3]环化反应[33-35]。反应以二价铜为催化剂,TEMPO 为助氧剂,实现了脒与酮的分子间氧化偶联反应,能够以较高的收率合成一系列嘧啶衍生物。该反应表现出了良好的官能团耐受性,为多种嘧啶类和喹唑啉类化合物提供了一种可行途径。通过对机理的研究,提出了可能的反应机理,首先,脒在碱的作用下与酮羰基脱水缩合成生成亚胺中间体15A,互变异构形成乙烯基亚胺15B。然后,15B 中与烯烃相连的甲基容易失去质子,进而与二价铜盐发生阴离子交换生成铜的碳复合物15C,紧接着均裂产生碳自由基15D 和一价铜,铜被氧化成二价铜继续参与循环。随后15D 被TEMPO 捕获随后消除形成不饱和醛,C-O 键氧化裂解得到羰基中间体15F。最后,中间体在氧气气氛下环化生成最终产物35a(图15)。
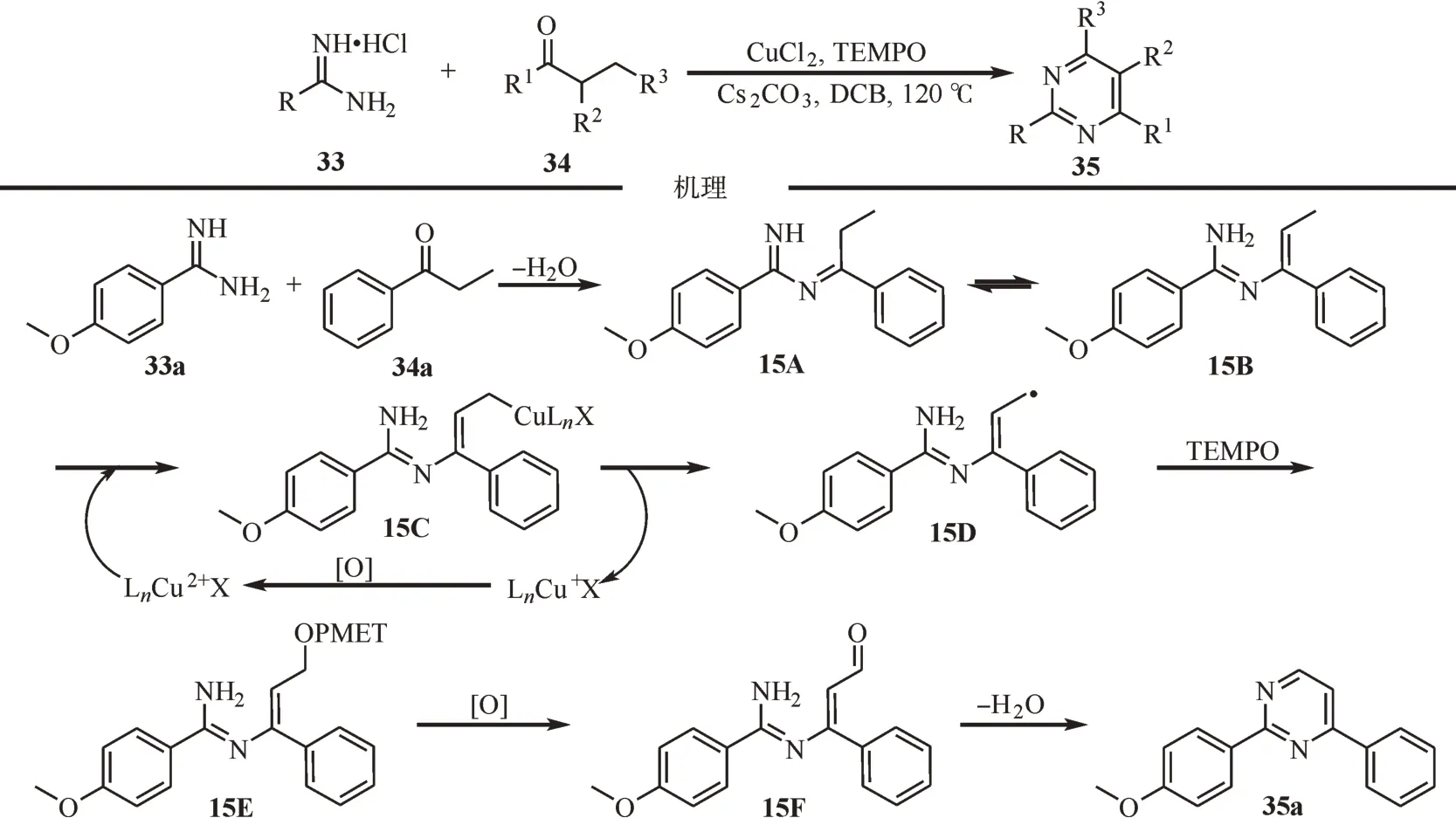
图15 铜催化脒盐和丙酮类衍生物参与的[3+3]环化反应Fig.15 Copper-catalyzed[3+3]cyclization reactions of amidine salts with acetone derivatives
2017 年,毕锡和等报道了一种可见光条件下活性亚甲基、全氟烷基碘化物和脒参与的[2+1+3]环化反应,为全氟烷基嘧啶提供了一种模块式合成新方法[36-37]。反应以氢氧化钠作碱,乙腈为溶剂,在室温的条件下就能以中等到优良的收率得到目标产物。基于控制实验,反应的可能过程为,首先在碱的作用下,β-酮酸酯烯醇互变后被夺取质子得到烯醇化物16A 作为卤键受体,全氟丁基碘作为卤键供体,两者相互作用原位形成卤键配合物16B。配合物16B 在可见光作用下经过单电子转移过程生成碳自由基和全氟丁基碘自由基阴离子,其释放碘离子以及产生C4F9自由基。然后,16C 和C4F9自由基之间的自由基交叉偶联产生α-全氟烷基化中间体16D,经过消除HF 形成烯烃16E。接着脒或胍亲核试剂与烯烃发生迈克尔加成再脱除HF 得到烯烃16F,16F 可以通过烯醇互变将乙酰基移位生成中间体16G。16G 最后发生分子内缩合脱水产生最终产物39a(图16)。

图16 可见光诱导的活性亚甲基、全氟烷基碘化物和脒盐的串联环化反应Fig.16 Visible light-induced tandem cyclization reactions of reactive methylene,perfluoroalkyl iodide and amidine salts
2018 年,沈志良等报道了1 例在铜催化下脒盐、苯乙烯和氟烷基卤化物参与的三组分环化反应,该反应可以在一锅中形成3 个新的C-C/C-N键和1 个新的六元环,为构建嘧啶衍生物提供了一种新路径[38]。结合自由基捕获等控制实验,提出了一个可能机理路径:首先,通过铜盐诱导TBHP均裂得到t-BuO 和t-BuOO 自由基。同时,一价铜催化的全氟丁基碘的C-I 键断裂释放出亲电子的n-C4F9自由基17A。随后,将新生成的全氟烷基自由基加成到烯烃的C=C 上,形成活性苄基中间体17B。而该中间体17B 又易被过氧化氢叔丁基氧化生成1 个短暂存在的中间体17C。α-全氟烷基化酮17C 在DABCO 作用下可以消除1 个HF 分子,得到脱氟中间体17D。最后,含双亲电位点的α,β-不饱和酮17D 与含亲核位点的脒发生电子匹配的[3+3]环化,接着发生β-F 消除产生全氟烷基化嘧啶43a(图17)。
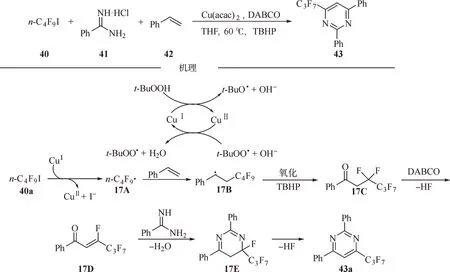
图17 铜催化脒盐、苯乙烯和氟烷基卤化物参与的三组分串联环化反应Fig.17 Copper-catalyzed three-component tandem cyclization reactions of amidine salts,styrene and fluoroalkyl halides
3 总结与展望
综上所述,本文总结了2015 年以来脒盐参与的串联环化反应及在构建含氮杂环方面的应用进展,主要从无过渡金属体系和自由基型反应两个方面对其进行了归纳概述。同时,着重对反应策略和反应机理进行了详细分析,可望为脒类的后续反应设计提供系统性的参考。可见光催化合成与传统合成方法相比,具有相当大的优势,即催化剂的可循环性好、条件温和、绿色高效等优点,然而注意到只有少数脒盐相关的反应利用了光催化的反应条件。电催化由于具有选择性高、副产物少、反应温和、官能团耐受性好等优点,近些年也取得了飞速的发展,但涉及脒盐的相关反应例证还是非常有限。可以预见通过进一步将可见光催化合成或电催化合成与脒盐化学结合起来,探究其在含氮杂环化合物上的合成应用也是未来值得深入探索的领域。由于这些含氮杂环类化合物在小分子药物领域的突出价值和其它应用功能,我们相信脒盐相关反应会越来越多地被应用于医药和农药研发中,乃至材料及催化等科研领域。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
汕头大学学报(自然科学版)(2020年4期)2020-12-14
造纸化学品(2017年1期)2017-01-21
化工生产与技术(2016年5期)2016-03-13
化工生产与技术(2016年5期)2016-03-13
中国塑料(2015年6期)2015-11-13
股市动态分析(2015年12期)2015-09-10
分析化学(2015年7期)2015-07-30
中国当代医药(2015年7期)2015-03-01
无机化学学报(2014年12期)2014-02-28
