
Low temperature conversion of methane to syngas using lattice oxygen over NiO-MgO
2022-11-05 06:47JunuWangZeaiHuangYingWangJundaoWuZhiqiangRaoFangWangYingZhou
Chinese Chemical Letters 2022年10期
Junu Wang,Zeai Huang,*,Ying Wang,Jundao Wu,Zhiqiang Rao,Fang Wang,Ying Zhou,*
a State Key Laboratory of Oil and Gas Reservoir Geology and Exploitation,Southwest Petroleum University,Chengdu 610500,China
b Institute of Carbon neutrality &School of New Energy and Materials,Southwest Petroleum University,Chengdu 610500,China
Keywords:Methane Lattice oxygen Syngas Hydrogen NiO
ABSTRACT The conversion of methane to syngas (H2 and CO) is an important route to produce high value-added products.Oxidize methane into syngas in the absence of gaseous oxidants is an economical route.In this work,NiO-MgO composite is successfully synthesized via an impregnation method.At 764 K,methane is directly converted to syngas on the NiO-MgO without gaseous oxidants.A synergistic effect of NiO and MgO was observed,in which NiO induced lattice oxygen of MgO mobility to oxidize methane and suppressed the formation of intermediates for side reaction.As a result,NiO-MgO exhibited enhancement of catalytic activity with the H2 production rate of 1241.0 μmol g-1 min-1,which was 3.4 times higher than that of pure MgO.This work provides a direct guidance to understand of methane oxidation via lattice oxygen under low temperature (<773 K).
Methane as the main component of natural gas is abundantly available throughout the world [1,2].As the cleanest fossil fuel,methane conversion and utilization as an alternative of petrochemical industry has aroused intense interests [3].The conversion of methane to syngas (H2and CO) is an important route for the production of high value-added productsviathe Fischer-Tropsch synthesis [4].Nevertheless,traditional dry/steam reforming methane to obtain syngas is an endothermic progress with high energy input and various side reactions [5,6].In contrast,methane partial oxidation is an exothermic reaction [7],but such process requires pure oxygen,leading to the drawbacks of explosion risk,additional cost and excessive oxidation.Alternatively,methane conversion to syngas using lattice oxygen of metal oxide catalysts (oxygen carrier) could avoid direct contact with oxygen gas.The used catalysts can be re-oxidized with different oxidants such as CO2,H2O and O2through chemical looping process [8].Therefore,methane conversionviaoxygen carrier is an attractive route for cost-effective commodity chemical and energy-effective production [9,10].
In general,methane requires high energy to activate due to the high energy barrier for C-H bond breaking [11-13].Therefore,high temperature (>1173 K) is often necessary to trigger the methane conversion.Recent studies rarely achieved methane oxidation under temperature less than 1173 K [14-16].Besides it is difficult to prevent the successive cleavage of C-H bonds under high temperature conditions,leading to the carbon deposition and catalyst deactivation [17].It is highly desired but of great challenge to develop catalysts with high efficiency at low temperature (<773 K) [18].
At these points,it is critical to design catalysts which could both raise methane conversion and reduce the reaction temperature.Non-noble nickel based catalysts have been proved to activate methane and enhance the lattice oxygen mobility from metal oxide substrates at low temperatures [19,20].The Ni-oxide interface is believed to facilitate methane activation with a partially positive Ni oxidation state to assist the cleavage of the C-H bond [21,22].In addition,C-H bond activation barriers relate closely to the extent of Ni-to-CH3interactions [23,24].However,metallic Ni usually showed strong cleavage of C-H bonds,leading to complete decomposition of methane and deactivated catalysts.This fact poses the great challenges of inhibiting the reduction of nickel oxide to maintain stable oxidation of methane.
MgO was used as the support due to the high oxygen storage capacities to donate its oxygen [25-28].It has been reported that MgO as a support significantly promotes reaction rate as well as the oxygen utilization,such as red mud with MgO improved the cyclic stability methane [12],MgO/Fe2O3increased lattice oxygen utilized [29]and Ce-Fe-Zr-O/MgO formed a porous interface layer to promote the methane conversion [11].However,these binary-MgO catalysts were normally inactive at 773 K or lower.Inspired by the chemical looping reforming of methane which can oxidize CH4to syngas,we designed NiO modified MgO oxygen carrier catalyst.The resultant NiO-MgO is highly active to convert CH4at temperature of 764 K.It was found that NiO-MgO was an effi-cient catalyst for methane oxidation to CO,and the lost oxygen of NiO was supplied by lattice oxygen of MgO.Particularly,the 1241.0 μmol g-1min-1H2yield can be achieved in a synergetic combination of NiO and MgO.The key intermediate species for side reaction was significantly decreased on NiO-MgO as compared to that of pristine MgO during methane conversion.This work provides a route for methane specific catalytic transformationsvialattice oxygen under low temperature.

Fig.1.(a) TEM image of NiO-MgO;(b) HRTEM image of NiO-MgO;(c-g) STEM and EDS mapping of NiO-MgO.
In this work,impregnation of Ni precursor into MgO led to the formation of NiO-MgO metal-support interaction after calcination.The loading of Ni was 9.7 wt% (Table S1 in Supporting information).X-ray diffraction (XRD) patterns of MgO and NiO modified MgO showed no obvious changes,and careful analysis could be seen slight peak shifts at 2θ= 42.9°,62.3°,78.9° (Fig.S1 in Supporting information),which was due to that both NiO and MgO were agglomerated with overlap of each other.Such similar crystal structure is believed to be beneficial for the formation of strong interfacial interaction [30].No peaks of metallic Ni were observed in XRD.Small particles uniformly distributing on the surface of the MgO nanoparticles were observed as shown in both transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images (Figs.1a-c and Figs.S2a and b in Supporting information).The high resolution TEM (HRTEM) image of NiO-MgO showed that there was an obvious interfacial contact between NiO and MgO (Fig.1b).Besides,the corresponding selected energy-dispersive spectrometry (EDS) mapping (Figs.1d-g) reconfirmed these nanoparticles were evenly distributed on the MgO surface.Moreover,strong interfacial interaction of NiO-MgO was evaluated by H2-temperature programmed reduction (H2-TPR) (Fig.S3 in Supporting information).As for NiO-MgO,the H2-TPR profile presented three obvious reduction peaks approaching at 696 K,802 K and 1045 K,which were corresponding to the reduction of NiO,NiO with weak interaction of MgO,and strong interaction of MgO,respectively [31-33].
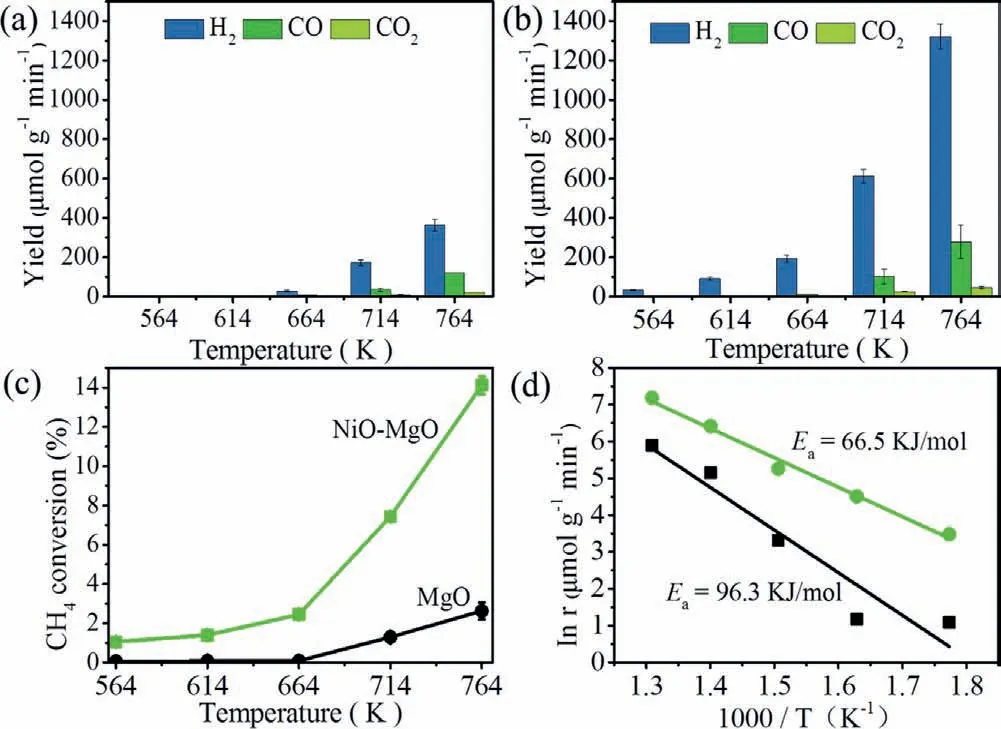
Fig.2.(a) Yield of H2,CO and CO2 over MgO;(b) yield of H2,CO and CO2 over NiOMgO;(c) CH4 conversion;(d) Arrhenius plots of apparent activation barriers for H2 formation over MgO and NiO-MgO (feed: 20% CH4/Ar,20 mL/min).
The methane conversion activities using oxygen carrier over MgO and NiO-MgO catalysts were shown in Figs.2a and b.The products of H2and CO increased with increasing the reaction temperature.NiO-MgO showed activity at 564 K,which was 100 K lower than that of MgO (664 K).At 764 K,the H2yield of NiO-MgO was 1241.0 μmol g-1min-1,which was a 3.4-fold enhancement of MgO (364.2 μmol g-1min-1).The CH4conversion efficiency over NiO-MgO was 14.1% at 764 K (Fig.2c),which showed well activity and selectivity in methane conversion to syngas using lattice oxygen under low temperature as compared to recent works (Table S2 in Supporting information).The corresponding Arrhenius plots (Ea,Fig.2d) of the NiO-MgO(66.5 kJ/mol) was significantly lower compared with MgO(96.3 kJ/mol),suggesting that the reaction barriers of methane activation should be reduced by NiO-MgO.Simultaneously,in the range of 564-764 K,it was interesting to note that the yield of H2O(MS signal:m/z= 18) was almost equal between MgO and NiOMgO (Figs.S4a and b in Supporting information),implying that the lattice oxygen over NiO-MgO was combined with carbon to form CO instead of forming water.
For a better understanding of the reduction progress of MgO before and after NiO modification,electron paramagnetic resonance (EPR) results showed that the concentration of oxygen vacancies in NiO-MgO atg= 2.003 was much higher than that of MgO (Fig.S5 in Supporting information).The increased oxygen vacancy density was believed to be responsible for the enhancement of lattice oxygen mobility.Such phenomenon was also confirmed by monitoring H2reaction usingin situdiffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) (Figs.S6a and b in Supporting information).The peak at around 1350 cm-1assigned to the surface lattice oxygen gradually weakened and disappeared significantly on NiO-MgO surface with the increasing of temperature [34].In contrast,there was no obvious peak changed on MgO.The above results indicated that modification of NiO on MgO enhanced the oxygen availability and mobility.
To understand the lattice oxygen changes of MgO and NiOMgO before and after reaction,X-ray photoelectron spectroscopy(XPS) of O 1s was studied (Fig.3a).The O 1s peak was divided into two peaks in all samples.The peak at binding energy of 529.6 eV was assigned to surface lattice oxygen (Olatt) and the peak at binding energy of 531.5 eV was assigned to surface absorbed oxygen (Oads) [35,36].The Olatt/Oadsafter NiO modification(Olatt/Oads= 1.9) showed an obvious increase as compared to bare MgO (Olatt/Oads= 1.6) (Table S3 in Supporting information),indicating that NiO-MgO could provide more lattice oxygen for methane oxidation.After reaction,the O 1s binding energy of both samples shifted to higher energy,indicating relatively lower electron attraction near the surface of the oxygen atoms as compared with samples before reaction [37].
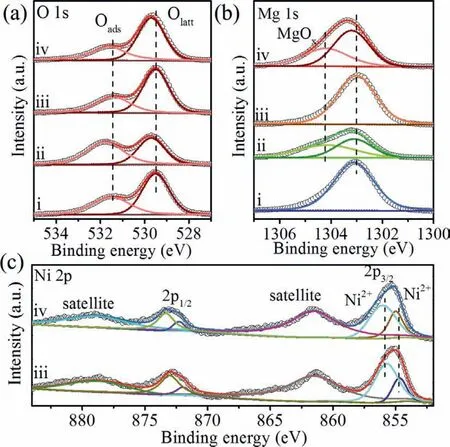
Fig.3.XPS spectra of MgO and NiO-MgO: (a) O 1s,(b) Mg 1s,and (c) Ni 2p (i:fresh MgO,ii: used MgO,iii: fresh NiO-MgO,iv: used NiO-MgO).
Furthermore,the Olatt/Oadsof MgO decreased obviously from 1.6 of sample before reaction to 0.8 of sample after reaction,indicating that the consumption of lattice oxygen from MgO during reaction.Interestingly,the Olatt/Oadsof NiO-MgO showed maintained at 1.9 before and after reaction,this should be due to the supply of oxygen from MgO to NiO during reaction.Firstly,the peak of Mg 1s after reaction could be divided to be MgOxat binding energy of 1304.3 eV and magnesium ion at 1303.2 eV,respectively [38-40].Such phenomenon should be due to the consumption of lattice oxygen from MgO,because Mg 1s of both samples showed no obvious peak at 1304.3 eV (Fig.3b).Secondly,Ni 2p peaks showed no obvious changes before and after reaction (Fig.3c),indicating that NiO maintained a stable state during reaction,further confirmed the lattice oxygen consumed should be from MgO in both MgO and NiO-MgO.Therefore,the activation of methane over NiO-MgO should be followed by the Mars-van Krevelen (MVK) mechanism[41,42].NiO as the active species during methane oxidation underwent NiO-Ni-NiO cycling process and the MgO support as the lattice oxygen supply to make the reaction continually going.
In situDRIFTS under CH4following with different temperature over MgO and NiO-MgO was performed to investigate the reaction mechanism of methane using surface oxygen carriers.Both MgO and NiO-MgO showed obvious-CH3adsorption at around 1340 cm-1from 298 to 764 K.However,the intermediates showed significant differences between MgO and NiO-MgO.CO was formed at low temperature as low as 364 K over NiO-MgO (Fig.4a),evidenced by the CO bonded with NiO at 2209 cm-1and 2239 cm-1assigned to CO species weakly bond to surface Ni atoms (Ni2+-CO)[43,44].CO adsorbed on NiO were believed to be easily desorbed as compared to that on MgO [45].These Ni2+-CO species changed to be gaseous CO (2182 and 2116 cm-1) when the reaction temperature was higher than 664 K.However,only gaseous CO was found from 414 K over MgO,indicating that NiO-MgO could trigger the low temperature methane oxidation to CO as compared to MgO.
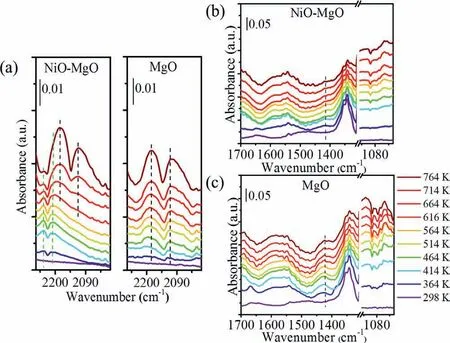
Fig.4.In situ DRIFTS spectra for MgO and NiO-MgO: (a) 2270-2000 cm-1 of two catalysts,(b) 1700-1000 cm-1 of NiO-MgO catalyst,and (c) 1700-1000 cm-1 of MgO catalysts,as the temperature was raised from 364 K to 764 K,under 20%CH4/Ar.
It is proposed that formation of Ni2+-CO on NiO-MgO was a key step for low temperature methane oxidation to syngas.Methoxy group (CHxO,around 1000-1150 cm-1) [46],bicarbonate (HCO3-,around 1430 cm-1) [47],and carbonate (CO32-,around 1550 cm-1)[48]species were formed over MgO at temperature higher than 364 K,these peaks showed an obvious increase when the temperature was increased to 764 K (Figs.4b and c).However,these intermediate species were significantly suppressed on surface of NiO-MgO,CHxO species were formed at 464 K,HCO3-species almost disappeared from 464 K.In combination with the formation of Ni2+-CO at lower temperature of 364 K,it is possible that CO was directly formed after CH4was cleavage over NiO-MgO.Besides,these species were also believed to be important intermediates for the formation of CH4[49].Indicated that the reverse reaction for CH4formation could be suppressed on NiO-MgO.
Based on the above analysis,the mechanism of methane conversion to syngas over NiO-MgO was shown at low temperature.In summary,an efficient catalytic conversion of CH4with a H2production rate of 1241.0 μmol g-1min-1was undertaken over NiOMgO at 764 K.Moreover,strong interfacial interaction effect,favorable oxygen availability and mobility,as well as less intermediates through the directly formation of Ni2+-CO route on NiO-MgO promote the conversion of CH4,which ultimately boost the effi-cient production of H2and CO.This discovery may provide a direct route to promote the low temperature methane conversion without gaseous oxidants.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgment
This research was financially supported by the Sichuan Provincial International Cooperation Project,China (Nos.2019YFH0164 and 2021YFH0055).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.12.060.
 Chinese Chemical Letters2022年10期
Chinese Chemical Letters2022年10期
- Chinese Chemical Letters的其它文章
- An odyssey of lithium metal anode in liquid lithium-sulfur batteries
- Recent progress on preparation and applications of layered double hydroxides
- Two-dimensional transition metal chalcogenide nanomaterials for cancer diagnosis and treatment
- Emerging nanomedicine and prodrug delivery strategies for the treatment of inflammatory bowel disease
- Recent advances in persulfate-based advanced oxidation processes for organic wastewater treatment
- Recent advance of fluorescent probes for detection of drug-induced liver injury markers