
Nanoparticles targeting at methylases with high correlation to N6-methyladenosine-related lncRNA signatures as potential therapy of kidney clear cell carcinoma
2022-11-05 06:47RuixunChnPingOuyngLicongSuXiXuPnghuLinYnqinLiQiGoYifnZhngShngNiFnLuoRuqiXuXiodongZhngXioxiLiYuCoPiynGoJunjunKngJunWuLuLi
Chinese Chemical Letters 2022年10期
Ruixun Chn,Ping Ouyng,Licong Su,Xi Xu,Pnghu Lin,Ynqin Li,Qi Go,Yifn Zhng,Shng Ni,Fn Luo,Ruqi Xu,Xiodong Zhng,Xioxi Li,Yu Co,Piyn Go,Junjun Kng,Jun Wu,Lu Li
a National Clinical Research Center for Kidney Disease,State Key Laboratory of Organ Failure Research,Guangdong Provincial Clinical Research Center for Kidney Disease,Nanfang Hospital,Southern Medical University,Guangzhou 510000,China
b Department of Health Management,Nanfang Hospital,Southern Medical University,Guangzhou 510000,China
c Department of Hematology,Nanfang Hospital,Southern Medical University,Guangzhou 510000,China
d Department of Urology,Peking Union Medical College Hospital,Chinese Academy of Medical Sciences and Peking Union Medical College (CAMS &PUMC),Beijing 100730,China
e Department of Urology,Nanfang Hospital,Southern Medical University,Guangzhou 510000,China
f Affiliated Foshan Maternity &Child Healthcare Hospital,Southern Medical University,Foshan 528000,China
g School of Biomedical Engineering,Sun Yat-sen University,Shenzhen 518107,China
h Department of Radiation Oncology,Nanfang Hospital,Southern Medical University,Guangzhou 510000,China
Keywords:Nanoparticle Methylation Long noncoding RNA Clear cell renal carcinoma Immunotherapy
ABSTRACT Clear cell renal cell carcinoma (ccRCC) is a heterogeneous malignancy with poor prognosis.Methylation of the N6 position of adenosine (m6A),the most common epigenetic modification in both messenger RNAs and noncoding RNAs,has been reported to regulate the initiation and progression of ccRCC.However,whether and how m6A-related long noncoding RNAs (m6ArlncRNAs) signify the progression of ccRCC remain unclear.We found m6ArlncRNAs are effective signatures illustrating immune landscape and risk stratification in ccRCC.We identified two differently expressed m6ArlncRNAs (DEm6ArlncRNAs),AC008870.2 and EMX2OS,as independent risk factors for overall survival of ccRCC patients,by applying stringent variable selection procedure to data from the Cancer Genome Atlas Kidney Renal Clear Cell Carcinoma project.The risk score generated from the DEm6ArlncRNA expression categorizes patients into either high or low-risk groups,between which,enrichment analysis indicated an enrichment in immunerelated pathways.Under different DEm6ArlncRNA transcription pattern,the two risk groups differ in immune cell population composition and expression levels of therapy targeting genes.Nanoparticle is satisfactory strategy to delivering therapeutic drugs.For further clinical translation,we designed a novel nanoparticle delivery system packaged STM2457 (STM@8P4 NPs),which selectively inhibits AC008870.2-correlated m6A writer.STM@8P4 NPs loaded drug successfully with uniform particle size,long-term stability and high release efficiency.STM@8P4 NPs can easily enter ccRCC cells and showed a highly efficient ccRCC killing activity in vitro.Our results therefore indicate that m6ArlncRNAs expression can depict tumor microenvironment,predict prognosis for ccRCC patient and give hint to therapeutic strategies in ccRCC.
Renal cell carcinoma (RCC) is a common and deadly malignancy,affecting approximately 175,000 people worldwide in 2018 and comprising a spectrum of highly heterogeneous subtypes [1].Clear cell RCC (ccRCC),the major (60%-80%) histological subtype,has a poor prognosis and responds weakly to radiotherapy and chemotherapy [2].However,with application of immunotherapy and targeted therapies,the prognosis of ccRCC patients has been greatly improved.In the wake of innovative therapeutic tools developing and molecular-level differences between various ccRCC subtypes being described,individualized decision making in ccRCC patients is becoming increasingly complex,calling for frontline molecular or genomic hallmarks for prognosis predicting and treatment selecting.
Currently,the available ccRCC risk models,such as Memorial Slogan Kettering Cancer Center (MSKCC) and the International Metastatic RCC Database Consortium (IMDC),become inaccurate to predict patient outcome or guide therapy,especially with newly approved tyrosine kinase inhibitors (TKI) plus immune checkpoint inhibitors (ICI) for ccRCC treatments by Food and Drug Administration.These risk models only integrate clinical and laboratory parameters roughly to classify patients into favorable-outcome,intermediate-outcome,and poor-outcome groups,among which the combination therapy of TKI and ICI shows similar efficacy [3].However,recent studies revealed the high potential of generated signatures of angiogenesis and immune infiltration based on transcriptional profile of ccRCC in predicting the clinical outcomes[4-6].For example,Senbabaogluet al.[4]dissected transcription profile to identify a macrophage-enriched ccRCC subtype,TCb,which was found to be associated with the lowest survival rate.Such results illustrated that an underlying molecular pathophysiological mechanism may explain ccRCC heterogeneity,thus suggesting RNA expression-based features need to be included and optimized in the ccRCC risk models for subclassifying ccRCC,predicting therapy response and delineating tumor microenvironments(TMEs).
m6A is the most common RNA modification in mammals which participates in almost every aspect of RNA metabolism (including transcription,translation,and degradation [7]) and regulated by a growing list of proteins (including m6A ‘writers’,‘readers’,and‘erasers’).A special subset of long noncoding RNAs (lncRNAs) can be modified by methylation at the N6 position of adenosine (m6A).These m6A-related lncRNAs (m6ArlncRNAs) with RNA expressionbased features are tightly linked to two unique features of ccRCC:angiogenesis (by influencing the cell cycle of myeloid cells) and immune-cell infiltration (by mediating immune-cell transcription)[8],thus may serve as important indicators to improve performance of ccRCC risk models.
Recently,regulating activity at m6A modifiers were reported as potential therapeutic strategy with effectively reducing growth of renal carcinoma [9,10].Parallel efforts to form small molecules targeting at m6A regulatory proteins (e.g.,STM2457 silencing methylase METTL3 [11]or FB23 suppressing demethylase FTO [12]) highlight the therapeutic potential of controlling RNA methylation in acute myeloid leukemia.
Nanobiotechnology as a novel strategy of targeted treatment in cancer has provided numerous nanoscale platforms for selectively delivering drugs [13,14]and enhancing permeability and retention [15,16].The range of materials used for aggregation has been expanded,along with different drug action parameters[17-19].Nanoparticle (NP) system with drugs loaded reduce toxicity and improve drug transportation efficacy,eliciting a favorable anti-tumor effect both in neoplasm itself and the tumor microenvironment [20-23].NP’s contribution to the field of ccRCC has been left largely unexplored,with few studies focusing on traffic preference to kidney [24].
Our research aims to distinguish an underlying subgroup of m6ArlncRNAs,unravel their potential for signifying molecular landscape and predicting overall survival in ccRCC patients and engineer a nanoparticle platform,to which we combine drugs targeting at vital m6A regulators,providing more accurate classification,prognostic assessment and novel therapy strategy (Fig.1).
As shown in Fig.S1 (Supporting information),omics study was conducted to identify those m6ArlncRNA biomarkers in the prognosis of ccRCC patients and the landscape of ccRCC microenvironment.Molecular data including raw counts and fragments per kilobase per million mapped reads and corresponding clinical data of ccRCC patients were obtained from the Cancer Genome Atlas (TCGA) data portal for Kidney Renal Clear Cell Carcinoma(KIRC) project.Based on the literatures [25-28],the following 36 m6A RNA methylation regulators (m6A readers,writers,and erasers) were selected manually to recognize m6ArlncRNAs: 23 readers (TRMT112,ZCCHC4,NUDT21,CPSF6,CBLL1,SETD2,HNRNPC,RBMX,HNRNPA2B1,IGF2BP1,IGF2BP2,IGF2BP3,YTHDC1,YTHDF1,YTHDF2,YTHDF3,YTHDC2,SRSF3,SRSF10,XRN1,FMR1,NXF1,PRRC2A);10 writers (VIRMA,METTL3,METTL5,METTL14,WTAP,RBM15,RBM15B,METTL16,ZC3H13,PCIF1);and 3 erasers(FTO,ALKBH5,ALKBH3).Genome Reference Consortium Human Build 38 (GRCh38) were acquired from the GENCODE website(www.gencodegenes.org/) for annotating lncRNAs.Co-expression analysis was then performed between the 36 m6A RNA methylation regulators and 9862 lncRNAs in KIRC data sets.Any lncRNA that was co-expressed with an m6A reader,writer,or eraser with|Pearson correlation coefficients|>0.6 andPvalue<0.001 was defined as an m6ArlncRNA.To select m6ArlncRNAs that are differentially expressing in 530 tumor tissues compared with in 72 normal controls,the R package limma [29]was used with a selection cutoff of log fold change (logFC) as 1.2 with false discovery rate(FDR) as 0.001.
For assess risk level in ccRCC patients,we designed a stringent procedure to select risk factors and employed those prognostic DEm6ArlncRNA to calculate the DEm6ArlncRNA risk score for each tumor samples.Factors included in MSKCC and IMDC risk models were inputted to variable filter as initially screen of clinical features.A subset of clinical and laboratory measurements was selected from 519 patients in KIRC project,excluding those with 0-day follow-up time.Nineteen selected clinical variables (List S2 in Supporting information) and 153 DEm6ArlncRNAs were added into the random forest model for 1000 cycles.Next,those variables with the top 25% Gini coefficient values were recognized as essential in each circle and were retained and counted.Univariable Cox proportional model was performed on those variables with frequency>950 times to characterize prognostic risk factors,following three hierarchical multivariable Cox models in which Features significantly correlated with overall survival in all-factor Cox model were chosen.Model 1 was unadjusted with only prognostic DEm6ArlncRNAs included.Model 2 was built on prognostic DEm6ArlncRNAs and was adjusted by AJCC (American Joint Committee on Cancer) stage,age and M stage.Model 3 was adjusted for variables (as in model 2) as well as for T stage and serum platelet level.To further validate the prognostic value of DEm6ArlncRNA signatures,the prediction performance between traditional risk survival models for metastatic ccRCC,MSKCC and IMDC,with the two models including key DEm6ArlncRNAs was compared based on C-index and time-dependent AUC.Nomogram was illustrated to estimate 1-/3-/5-year overall survival probabilities based on model 2.Sensitivity analysis was also performed to detect the robustness of relationship within various age groups and in different AJCC stages.DEm6ArlncRNAs significantly related to the outcome were kept for calculating DEm6ArlncRNA risk score,based on the following formula (Eq.1):

in the formula,lncRNAiindicates theithexpression value of prognostic DEm6ArlncRNAs andβiindicates the coefficient oflncRNAi.The threshold of DEm6ArlncRNA risk score was determined as the maximal value of true positive minus false positive at every point of the 5-year ROC curve,classifying the ccRCC patients into either the high-risk or low-risk group.Kaplan-Meier curves validated the cutoff point.The relationship between DEm6ArlncRNA risk score and clinical features were examined by Spearman’s correlation test.
To distinguish the key biological processes discerning high-risk and low-risk group,gene expression was compared in 123 highrisk patients and 396 low-risk patients by using limma.Then,functional gene enrichment analysis as well as gene set enrichment analysis (GSEA) [30]were conducted according to Gene Ontology(GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG)pathways downloaded from MsigDB (www.gsea-msigdb.org/gsea/msigdb).To discover the difference in immune infiltration status in detail,gene set variation analysis (GSVA),a method that estimates a variation of the given gene sets [31],was performed with applying a list of immune gene sets (List S1 in Supporting information)manually generated from published literature [32].
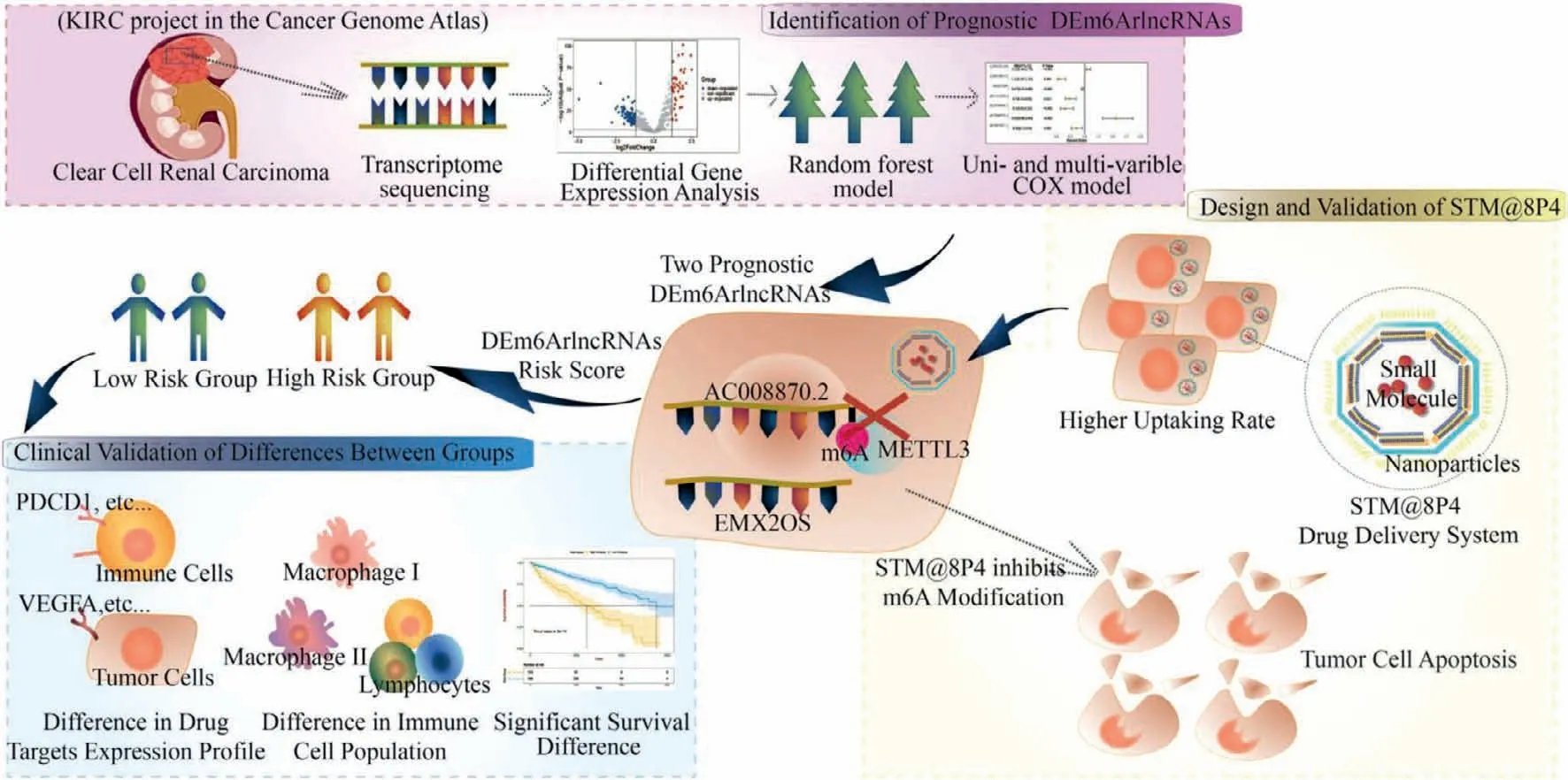
Fig.1.Illustrations depicting the three major parts of the design,including identification of prognostic DEm6ArlncRNA,clinical validation of differences between the two risk groups and design and validation of STM@8P4 NPs.
In investigation of tumor-infiltrating diversity between ccRCC risk groups,a variety of bioinformatic tools were applied to further analyze the association between DEm6ArlncRNA risk score and immune-infiltrating status.First,IPS and ESTIMATE functions were performed for delineating the immune-infiltrating status as a whole.IPS provided an evaluation of four main immunophenotypes and a generalized z-score [33],whereas ESTIMATE scored for the presence of stroma cells,the level of infiltrating immune cells,and the cellularity of tumor [5].Next,to evaluate the intratumoral immune-cell populations,a series of 7 immune-cell quantification tools,including XCELL,CIBERSORT,CIBERSORT.ABS,QUANTISEQ,MCPCOUNTER,EPIC and TIMIER,were applied by using TIMER 2.0 [34-38].These population estimates were then compared within high-risk group and low-risk group by using Wilcoxon test.Spearman’s rank correlation analysis was applied to further examine the association between DEm6ArlncRNA risk score and immune-cell populations.Further,we depicted the expression profile of molecules related to ICI and targeted therapies.The common agents targeted genes in ccRCC were manually integrated (List S3 in Supporting information).Wilcoxon test and Spearman’s rank correlation analysis were performed to investigate the relationship between the expression of targeted genes and the different level of m6ArlncRNAs risk score.
The synthesis procedure of STM@8P4 NPs was conducted by nanoprecipitation method.The formation of STM2457 has been detailed before [11].To prepare NPs,8P4 and STM2457 were dissolved in dimethyl sulfoxide and DSPE-PEG 2000,then we added the mixture drop into ultrapure water.Dynamic light scattering(DLS,Malvern Zetasizer Nano-ZS90) was applied for particle size measuring.In order to evaluate the drug loading and encapsulation rate of STM@8P4 NPs,the NPs were dispersed and diluted in DMSO.The concentration of STM2457 in NPs was calculated based on fluorescence spectrophotometry (Eqs.2 and 3).
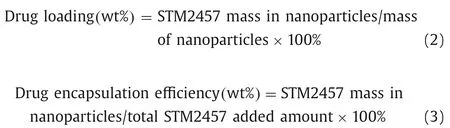
To test NPs’stability,NPs were introduced to 1×PBS and RPMI 1640 medium containing 10% FBS at room temperature for 7 days.Then,the particle size and polymer dispersity index (PDI) were measured at different time points with Zetasier Nano ZS90 for determination of long-term stability.A stability curve was also delineated with measured data.
Next,STM@8P4 NPs was sealed in dialysis bags (MWCO: 3500 Da).Then the dialysis bags were placed in different pH environments,including pH 5.0,6.8 and 7.4,which were created by the different volume ratios of citric acid and sodium hydrogen phosphate solutions.The system was put in a constant temperature shaker (37 °C,100 rpm).After that,at the specific time point (0.25,0.5,1,2,4,6,8,12,24,48,96 and 120 h),500 μL medium in the buffer solution was taken out for the measurement of STM2457 concentration,and the same volume buffer were added.The cumulative release of STM2457 was calculated at each time point.
For evaluate the cell uptake rate of STM@8P4 NPs,786-O cell lines were inoculated in a 15 mm glass-bottomed cell culture dish,cultured at 37 °C for 24 h then treated with Dil and Dil@8P4 for 0-8 h.The collected cells were stained by 4′,6-diamidino-2-phenylindole (DAPI) (1306,Thermo Scientific),fixed with 4%paraformaldehyde for 15 minutes,and observed through a highspeed confocal imaging system (Dragonfly CR-DFLY-202 2540).
To evaluate the cytotoxicityin vitro,786-O cell lines were selected and inoculated in a 15 mm glass-bottomed cell culture dish,cultured at 37 °C for 24 h,then treated with 8P4,STM2457,and STM@8P4 NPs for indicated hours.Cell proliferation fold changes were calculated by the ratio between the cell numbers at indicated time points 8P4,STM2457,and STM@8P4 NPs treatment compared to cells before treatment.For apoptosis analysis,cells were stained by AnnexinV (640907,Biolegend) and further incubated with 0.01 μg/μL DAPI (1306,Thermo Fisher Scientific) or 7-AAD (00699350,eBioscience) for 30 min at room temperature.IC50were calculated by the ratio between the cell numbers at 72 h with different concentrations of STM2457,and STM@8P4 NPs treatment compared to cells before treatment.The samples were analyzed using the Attune NxT flow cytometer (Thermo),and the results were analyzed using Flowjo software.All statistical analyses were performed on R platform (version 4.0.3)
As outlined above,in order to obtain DEm6ArlncRNAs in ccRCC patients,we attained transcriptome profiling data of 530 tumor samples and 72 adjacent normal tissues from TCGA KIRC project and annotated these data by using Ensembl ID (Fig.S1).We extracted a subgroup of 720 m6ArlncRNAs by conducting co-expression analysis between m6A related genes and lncRNAs.Among these m6ArlncRNAs,153 were recognized as DEm6ArlncRNAs in the tumor samples as compared to normal tissues using specific cutoff values (Figs.S2A-C in Supporting information).A subset of differentially-expressing transcripts were classified as m6ArlncRNA,indicating m6ArlncRNAs were linked to the occurrence of ccRCC.
To filter out important variables for ccRCC risk models,we used the random forest model with 1000 iterations to analyze the 153 DEm6ArlncRNAs in addition to 19 clinical features.Importantly,we found 12 variables that were essential for the prognosis of ccRCC,including 7 DEm6ArlncRNAs and 5 clinical features,namely AJCC stage,age,T stage,M stage,and serum platelet level.Next,we confirmed that all these variables significantly correlated with overall survival by utilizing univariate Cox proportional hazards model(Figs.2A-F).To further explore the independent risk factors for ccRCC overall survival and to calculate the prognostic index,we established three hierarchical multivariate Cox regression models.Our data demonstrated that two DEm6ArlncRNAs,AC008870.2 and EMX2OS,were independently associated with overall survival in all above-mentioned models (Table S1 in Supporting information).These correlations remained robust in the stratification of age,AJCC stage,and M stage,respectively (Table S2 in Supporting information).The corresponding nomogram was used to predict survival probability of the ccRCC patients with a 1-year AUC of 0.86(Figs.2G and H).Of note,adding these two DEm6ArlncRNA in both MSKCC based Cox model and IMDC based Cox model showed significantly improved C-index,1-year AUC,3-year AUC and 5-year AUC,suggesting a great advantage of combining DEm6ArlncRNA as prognostic factors when predict overall survival for ccRCC patients.(Table S3 in Supporting information).Two DEm6ArlncRNA pass the stringent variable selection procedure,through which we provided solid evidence that m6ArlncRNA expression in ccRCC patients were prognostically valuable.
We divided ccRCC patients into low-risk (n= 396) and highrisk (n= 123) groups based on DEm6ArlncRNA risk score formula built from model 2: DEm6ArlncRNA risk score=(0.482897×AC008870.2)+ (-0.057878× EMX2OS).The value 0.04618084 was set as the threshold to classify groups for evaluating their performance in the 5-year ROC.Compared to high-risk group,the overall survival was significantly longer in the patients of lowrisk group (Pvalue=6×10-14) (Fig.S3A in Supporting information).The Spearman correlation test revealed DEm6ArlncRNA risk score was highly related to clinical features that are prognostic predictors in IMDC or MSKCC models (Figs.S3B-G and S4A-D in Supporting information).The difference of clinical characteristics between these two groups were summarized in Table S4 (Supporting information).Patients with high-risk score had more advanced AJCC stage,higher serum platelet,and higher-grade pathological type than low-risk group (Pvalue<0.001).To further validate these differences,we performed limma analysis.We screened out 1133 upregulated genes and 470 downregulated genes between different risk groups (Fig.S5A in Supporting information).By performing KEGG and GO functional enrichment analysis,we found that immune-related pathways were activated in the highrisk group (such as defense response to bacterium,humoral immune response,and inflammatory mediator regulation of transient receptor potential channels) (Figs.S5B and C in Supporting information).In contrast,acid transport and metabolism pathways were activated in the low-risk group (Figs.S6A and B in Supporting information).GSEA confirmed these results: high-risk associated genes were highly enriched in immune-regulated biological processes (including complement activation and humoral immune response),and low-risk associated genes were enriched in small molecule catabolic process and anion transport (Figs.S5D-I and S6C in Supporting information).Finally,GSVA demonstrated that there was a slight but general increase in immune-related gene sets among high-risk ccRCC patients (Fig.S6D in Supporting information).These results further unraveled a latent interaction between m6A modification on EMX2OS/AC008870.2 and the ccRCC prognosis,combined with a strong association between immune-related molecular change and EMX2OS/AC008870.2 expression,thereby suggesting m6ArlncRNAs are likely to serve as the novel land markers for ccRCC.
Since immune-related biological processes were most enriched,we used a series of immune-infiltrating evaluation tools to dissect TME differences between high-risk and low-risk groups.First,we calculated immunophenoscores (IPSs) to create a general index for the immune-infiltration landscape in both groups.Irrespective of their risk group classification,ccRCC tumors had high IPSs.Interestingly,although no significant variation was found in the IPS score,the separate component scores were higher in high-risk group for effector cells but lower for immunsuppressive cells and immune checkpoints,suggesting the possibility of m6AlncRNAs associated immune TMEs shifting relating to prognostic risk in ccRCC(Fig.S7A in Supporting information).Next,we used ESTIMATE[5]to examine the stroma and immune cells infiltrating states.In patients with higher m6AlncRNA risk scores,more stromal cells were found to surround the neoplasms.Consistent with the individual component scores,the high-risk ccRCC TMEs had more infiltrating immune cells and a more compacted tumor cellularity than low-risk ccRCC TMEs (Fig.S7B in Supporting information).Finally,we used a combination of seven absolute or relative immunecell quantification tools to find the specific abundant types of immune cells in low-risk and high-risk TMEs.Endothelial cells,neutrophils,hematopoietic stem cells,and non-regulatory CD4+T cells were enriched in low-risk TMEs,whereas B cells,fibroblasts,macrophages,NK cells,CD4+naïve cells,follicular helper T cells,Th1 cells,and Treg cells were enriched in high-risk TMEs (Table S5 in Supporting information).Intriguingly,results generated by different immune-cell quantification tools were highly consistent.In addition,we confirmed that patients with different DEm6ArlncRNA risk scores had different cell populations enrichment by performing Spearman’s correlation analysis (Fig.S7C in Supporting information).The m6ArlncRNA-based risk score was correlated with distinct immune-cell populations in the TME,linking miserable prognosis to extensive infiltration of T cells in the ccRCC TME,which is likely resulted from most CD8+T cells functionally impaired or stably expressing suppressive co-receptors [39].
Anti-angiogenesis agents and ICI are efficacious for treating ccRCC and the combination therapy was recommended by European Association of Urology for treating metastatic RCC [40].Accordingly,we wondered whether DEm6ArlncRNA risk score was correlated with the expression levels of genes that were targeted by different therapies.In addition to common signals (e.g.,mTOR)inside tumor cell,immune checkpoints (e.g.,CTLA-4 and PDCD1)on T cells and endothelial cell markers (e.g.,VEGFR) were taken into consideration in the meantime [41].The expression profiles extracted from bulk RNA seq were different between high and low risk groups: FLT1,KDR,FLT4,BRAF,KIT,TEK,NTRK2,FGFR2/3 and EGFR expression levels were higher in the low-risk group,whereas PDGFRA,RET,TYRO3,ROS1,PDCD1,and CTLA4 expression levels were higher in high-risk group (Table S6 in Supporting information).Again,we verified this correlation by performing Spearman’s rank correlation analysis (Fig.S7D in Supporting information).The T cell-inhibitory receptors that could be blocked by ICI,were positively linked to high-risk group,suggesting patients with higher risk score were more likely to have improved response to ICI but react silently for VEGFR or mTOR inhibitors.We proposed that m6ArlncRNA expression could be an important signature for guiding individualized therapy in ccRCC patients.
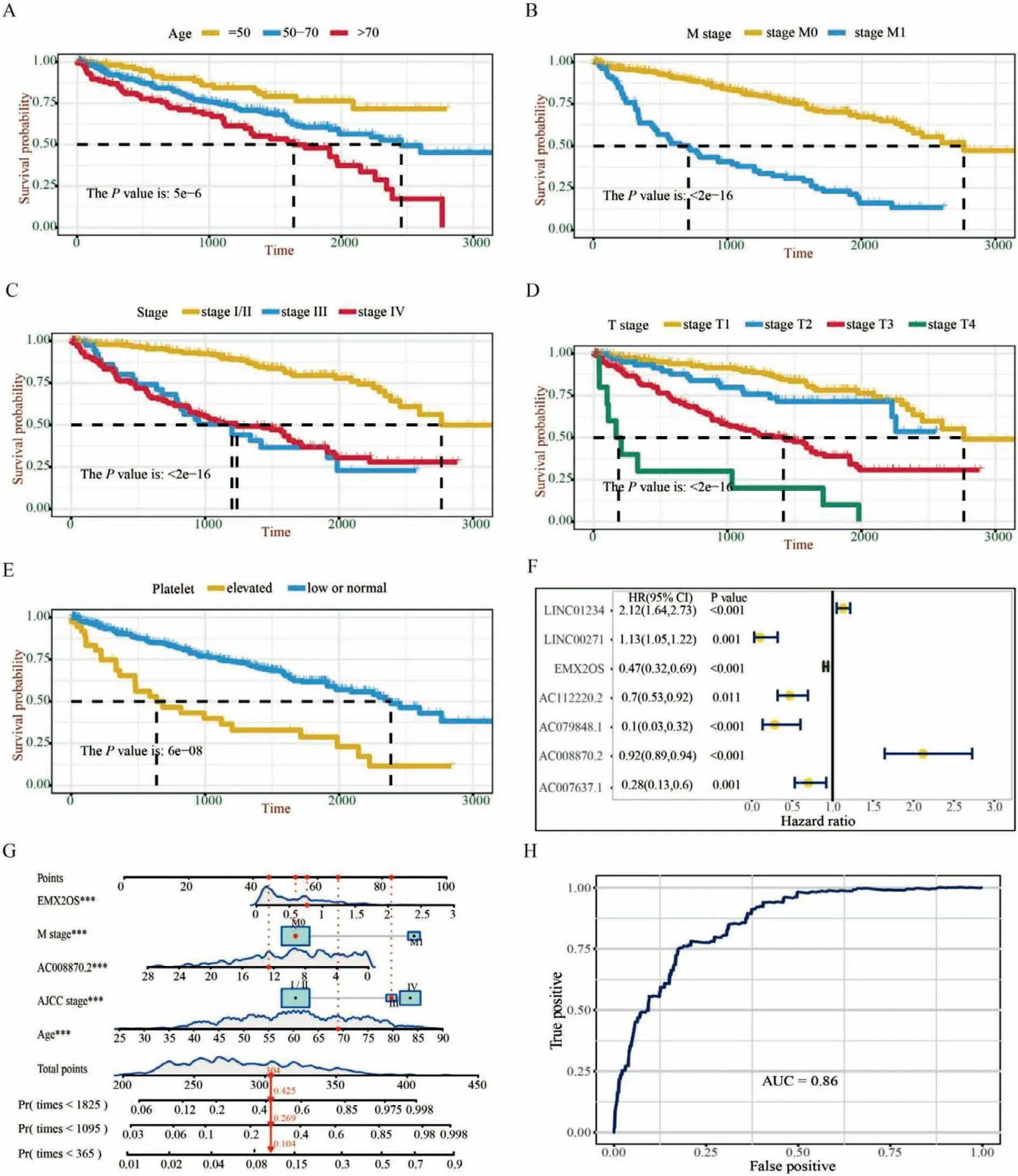
Fig.2.The selection of prognostic factors for ccRCC patients.(A-E) Kaplan-Meier curves showing that patients with different characteristics of selected clinical variables had different overall survival.(F) A forest map showed 7 selected m6ArlncRNAs after 1000 times random forest model.(G) A constructed nomogram for predicting overall survival in ccRCC patients.Red lines and dots show a specified patient’s point.(H) The AUC of nomogram model in predicting 1-year overall survival of ccRCC patients.
The analysis uncovered that m6ArlncRNAs not only predict overall survival of ccRCC patients but also signify the immune cell landscape in TME,combined with a strong correlation between their expression and key therapies-targeting molecules.The latent m6ArlncRNAs-based network inspired us with the therapeutic potential of interfering the m6A modifications on prognostic m6ArlncRNAs in ccRCC TME.Thus,we considered selective inhibitors targeting at those m6A regulators,loaded by nanoparticle drug delivery system for more efficient tumor locoregional pharmacokinetics.In order to investigate if blocking the selective m6A writer can bring a clinical translation as therapeutic strategy,we focused on AC008870.2,which was the risk factor in the two prognostic DEm6ArlncRNAs with the lowestPvalue.AC0088790.2 showed the highest positive correlation with methyltransteraselike 3 (METTL3),thus we employed STM2457,a selective inhibitor of METTL3/METTL14 complex identified by Yankovaet al.[11].For the purpose of improving the efficiency of STM2457 we here proposed 8P4 nanoparticles.The 8P4 nanoparticle platform has been successfully developed in solid tumor for enhancing tumour targeting efficiency of kinase inhibitors and chemotherapeutic drugs[42,43].The STM2457 was successfully packaged in 8P4 NPs as scheduled,and the average size of STM@8P4 NPs was 50.7 ± 1.05 nm with a narrow polydispersity index (Fig.3A).The drug loading and encapsulation efficiency of STM2457 in STM@8P4 NPs detected by fluorescence spectrophotometry were 6.9% ± 0.03% and 51.3% ± 0.15%,respectively (Table S7 in Supporting information).In thein vitrostability experiment,it was found that the particle size of STM@8P4 NPs did not change significantly after long-term storage,indicating that STM@8P4 NPs had good stability (Fig.3B).Considering that will transport through different pH environments in body,we incubated STM@8P4 NPs in a series of pH solutions to examine the release efficiency.The release of STM2457 at pH 5.0 was significantly higher than that at pH 7.4 (Fig.3C),suggesting that STM@8P4 NPs might rapidly release STM2457 in the acid tumor microenvironment but maintain a high concentration and long stability at physiological conditions.
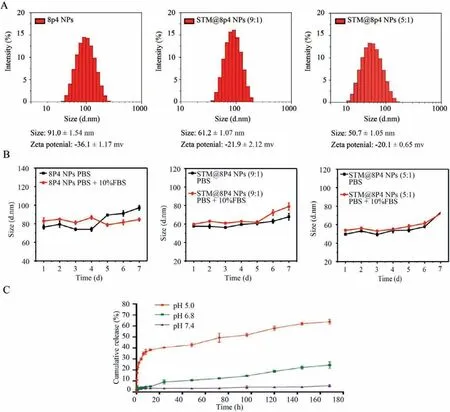
Fig.3.The characterization of STM@8P4 NPs.(A) Particle size distribution of 8L6 and STM@8P4 NPs.(B) Stability of STM@8P4 NPs.(C) Drug release behavior of STM@8P4 NPs in different pH solutions.
The uptake efficiency of chemotherapeutic drugs by tumor cells is an important factor in tumor killing efficiency.Therefore,in order to observe the uptake of 8P4 by renal clear cell carcinoma cells,we treated 786-O cell lines with Dil and Dil@8P4 to observe the intracellular drug content,found that 8P4 is very easily taken up by the 786-O cell line (Fig.4A,quantification in Fig.4B).To explore the therapeutic effects of STM@8P4 NPsin vitro,we treated 786-O renal clear cell carcinoma cells with 8P4,STM2457,and STM@8P4 NPs,respectively.Our data showed that STM@8P4 NPs efficiently inhibited 786-O cell proliferation more efficiently than STM2457 treatment at 72 h post-treatment (Figs.4C and D).Furthermore,STM@8P4 NPs induced more dramatic apoptosis in 786-O cells than STM2457 treatment (Fig.4E).In order to better study whether STM@8P4 NPs improve the toxicity of STM2457 renal clear cell carcinoma cells,we administered 0-106 nmol/L drug treatment to the 786-O cell line and found that STM@8P4 NPs can reduce the IC50of STM2457 against renal clear cell carcinoma cell lines from 6783 nmol/L to 98 nmol/L.
Taken together,m6ArlncRNAs are novel signatures of the immune landscape in ccRCC TME and overall survival in ccRCC patients,and by constructing the nano-biopolymer,it presents as an attractive anti-tumor potent to inhibit the m6A modifications on critical DEm6ArlncRNA AC008870.2.The nano-based drug delivery system,STP@8P4 NPs,to which we attached small molecule targeting METTL3,are stable and easily taken by ccRCC cells,generating a favorable pharmacokinetic pattern and further forming a remarkably better tumor elimination effect.In conclusion,we illustrate the potential of m6ArlncRNAs for guiding clinical therapy decisions and predicting prognosis and unravel that STM@8P4 NPs can serve as effective treatment of ccRCC by targeting at prognostic DEm6ArlncRNA related m6A modifiers.
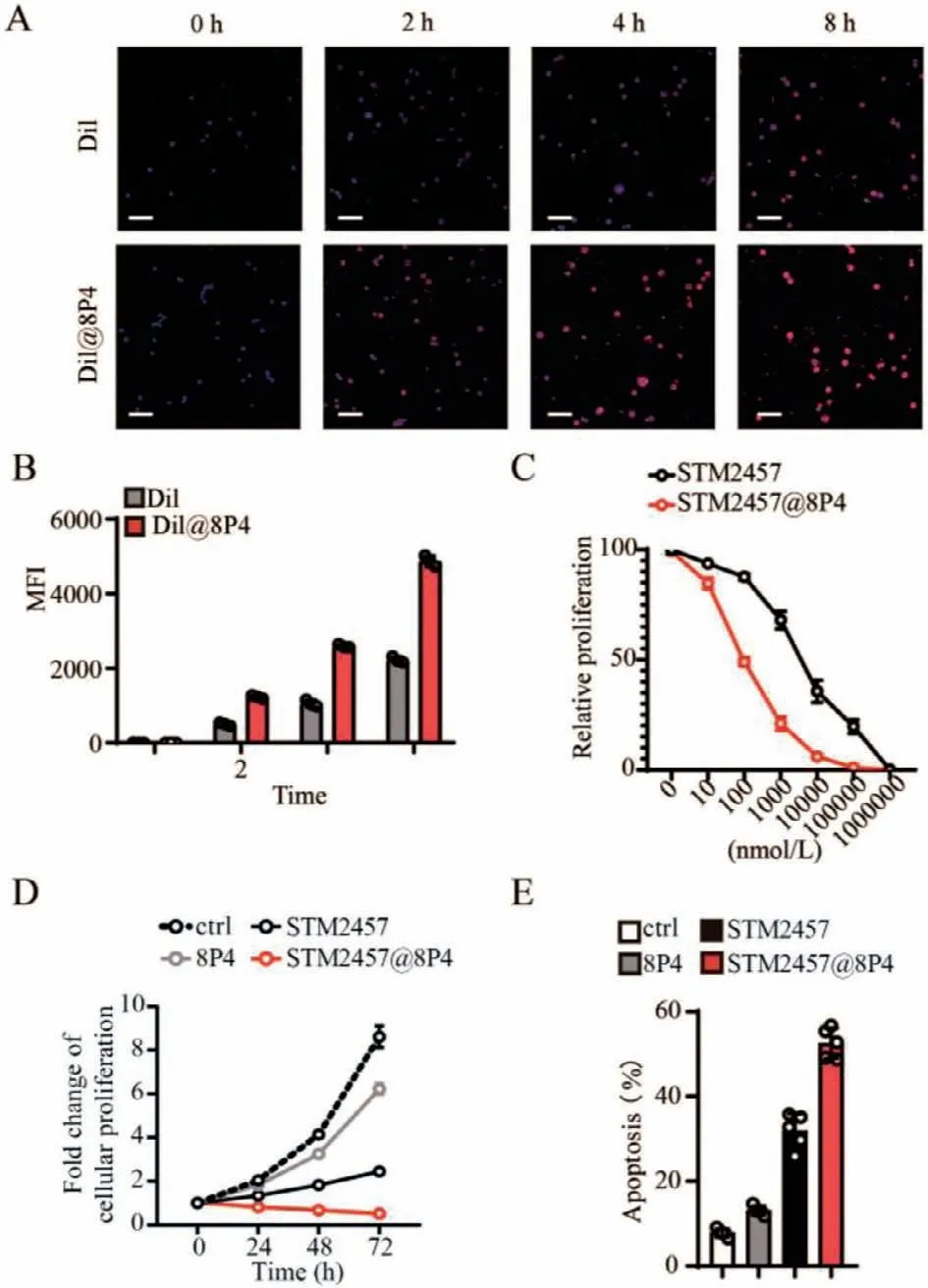
Fig.4.Pharmacological inhibition of STM@8P4 NPs in renal clear cell carcinoma cells.(A) Representative images and quantification (B) of Dil uptake in 786-O cells(Scale bar=20 μm).(C) Dose-response curves of a 786-O cells panel to STM2457 and STM@8P4 NPs.(D) The cell proliferation rate of 786-O cells incubated with PBS(Ctrl),8P4,STM2457,and STM@8P4 NPs (n=5 independent replicates per group).(E) Apoptotic 786-O cells at 72 h after indicated treatment (n=5 independent replicates per group).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This study was funded by the National Natural Science Foundation of China (Nos.8210102561,81900626,51973243,52173150),Nanfang Hospital (No.2019C028),International Cooperation and Exchange of the National Natural Science Foundation of China (No.51820105004),Science and Technology Planning Project of Shenzhen (No.JCYJ20190807155801657),Guangdong Innovative and Entrepreneurial Research Team Program (No.2016ZT0 6S029).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.03.074.
 Chinese Chemical Letters2022年10期
Chinese Chemical Letters2022年10期
- Chinese Chemical Letters的其它文章
- An odyssey of lithium metal anode in liquid lithium-sulfur batteries
- Recent progress on preparation and applications of layered double hydroxides
- Two-dimensional transition metal chalcogenide nanomaterials for cancer diagnosis and treatment
- Emerging nanomedicine and prodrug delivery strategies for the treatment of inflammatory bowel disease
- Recent advances in persulfate-based advanced oxidation processes for organic wastewater treatment
- Recent advance of fluorescent probes for detection of drug-induced liver injury markers