
QuEChERS-气相色谱—串联质谱法测定茶叶中水胺硫磷农药残留的不确定度评定
2022-11-02 01:08陆振华于丽芳蔡丹萍
食品与机械 2022年10期
陆振华 于丽芳 蔡丹萍 李 成
水胺硫磷是由德国拜耳公司研发的一种广谱性有机磷杀虫、杀螨剂,具触杀、杀卵和胃毒作用,在昆虫体内能首先被氧化成毒性更大的水胺氧磷,抑制昆虫体内的乙酰胆碱酯酶[1]。水胺硫磷属于高毒农药,已禁止在蔬菜、瓜果、茶叶、菌类、中草药材上使用。GB 2763—2021《食品安全国家标准 食品中农药最大残留限量》中规定茶叶中水胺硫磷的最大残留限量为0.05 mg/kg。近年来,茶叶中水胺硫磷超限量常有报道,如何提升农药残留检测结果的准确性已成为相关检测机构的一项重要工作。
目前,茶叶中农药残留的检测方法较多。对气相色谱法[2-4]、气相色谱—质谱法[4-6]和液相色谱—质谱法[7]测定茶叶中农药残留的不确定度评定有较多报道,关于GB 23200.113—2018《食品安全国家标准 植物源性食品中208种农药及其代谢物残留量的测定 气相色谱—质谱联用法》测定蔬菜中农药残留的不确定度评定也有报道[8-9],而参照GB 23200.113—2018测定茶叶中农药残留的不确定度评定还未见相关报道。茶叶基质复杂,含有较多的生物碱、茶多酚和色素等对农药残留检测产生干扰的物质,检测时需加入更多的净化试剂,且茶叶含水率较低,在提取过程中需用水浸泡使细胞膨胀,与有机溶剂充分接触,才能达到提高提取效率的目的。且茶叶农残检测时,取样量较少,称样量及均匀性对检测结果的影响较大。因此茶叶的前处理对检测结果的影响更大。对于各影响因素评定方法的处理,通常回收率有显著性差异时,会将回收率代入计算公式以修正样品的测定结果[10-11],但当实际出具的检测数据满足标准要求时,对回收率的处理并不直接折算到结果中。评定用内标法定量时的不确定度,通常的处理方法是用内标浓度进行评定再进行合成[12-13],但内标法一般用同一种内标,只与加标的体积有关。
研究基于CNAS-GL006—2019《化学分析中不确定度的评估指南》和JJF 1059.1—2012《测量不确定度评定与表示》,参照GB 23200.113—2018中QuEChERS-气相色谱—串联质谱法测定茶叶农药残留的方法,用单独的数学模型将内标参与到评定过程中,将回收率以纯度的方式参与到评定过程中,再对样品称量、体积引入、测量重复性等因素进行评定,找出影响茶叶中水胺硫磷检测结果测量不确定度的主要原因,以期为实验室检测茶叶中水胺硫磷时进一步提高检测数据的可靠性提供参考。
1 材料与方法
1.1 仪器与试剂
气相色谱质谱联用仪:GCMS-TQ8040型,日本SHIMADZU公司;
氮吹仪:N-EVAP 111型,美国Organomation公司;
旋涡仪:XH-B型,江苏康健医疗用品有限公司;
超声仪:KH-500DE型,昆山禾创超声仪器有限公司;
离心机:TG1850-WS型,上海卢湘仪离心机仪器有限公司;
电子天平:SL2002N型,上海民桥精密科学仪器有限公司;
乙腈、乙酸乙酯:色谱纯,美国Fisher公司;
水胺硫磷(CAS:24353-61-5;相对扩展不确定度±3%):1 000.0 μg/mL,北京曼哈格生物科技有限公司;
环氧七氯B(CAS:1024-57-3;相对扩展不确定度±3%):100.0 μg/mL,北京曼哈格生物科技有限公司;
乙二胺-N-丙基硅烷化硅胶(PSA):50 μm,纳谱分析技术(苏州)有限公司;
十八烷基硅烷键合硅胶(C18):50 μm,纳谱分析技术(苏州)有限公司;
石墨化炭黑(GCB):100 μm,纳谱分析技术(苏州)有限公司。
1.2 试验方法
1.2.1 试样处理
(1) 提取:准确称取2 g粉碎均匀的茶叶样品,于50 mL 塑料离心管中,加10 mL水涡旋混匀,静置30 min。加入15 mL乙腈—醋酸(V乙腈∶V醋酸=99∶1)溶液、6 g无水硫酸镁、1.5 g醋酸钠、1颗陶瓷均质子,涡旋1 min,超声30 min,4 200 r/min离心5 min。
(2) QuEChERS净化:吸取8 mL上清液到15 mL塑料离心管中(内含1 200 mg硫酸镁、400 mg PSA、400 mg C18及200 mg GCB),涡旋混匀1 min。4 200 r/min离心5 min。
(3) 浓缩:准确吸取2 mL上清液至10 mL玻璃试管中,40 ℃水浴中氮吹至近干。加入20 μL的内标溶液(环氧七氯B 5 μg/mL),加入1 mL乙酸乙酯复溶,过0.22 μm 滤膜,用于GC-MS/MS测定,内标法定量。
1.2.2 仪器检测条件
(1) 色谱条件:色谱柱为TG-5MS(30 m×0.25 mm×0.25 μm);流量1.69 mL/min;程序升温条件为初温50 ℃,保留1 min,以25 ℃/min升温至125 ℃,再以10 ℃/min 升温至300 ℃,保留6 min;进样口温度250 ℃;不分流进样,进样量体积1 μL。
(2) 质谱条件:EI源;电子轰击源70 eV;传输线温度250 ℃;离子源温度230 ℃;溶剂延迟2 min;多反应监测(MRM),优化后的质谱参数见表1。
1.2.3 测量数学模型
(1)
式中:
ω——待测样品中水胺硫磷的含量,mg/kg;
c——试样中水胺硫磷的质量浓度,μg/mL;
v——总的样品代表的试样体积,mL;
V1——加入提取溶剂的体积,mL;
V2——移取上清液的体积,mL;
V3——最终样品复溶体积,mL;
m——试样称样量,g;
fmethod——方法校正因子。
1.2.4 不确定度分量的主要来源 根据试验的测定,茶叶中水胺硫磷残留量测定不确定度分量主要来源如图1所示。

表1 水胺硫磷及环氧七氯B的质谱参数表Table 1 Mass spectrum parameters of isocarbophos and heptachlor epoxide B

图1 不确定度来源Figure 1 Analysis diagram of the uncertainty source
2 水胺硫磷农药残留不确定度分量的评定
2.1 样品称量引入的相对标准不确定度urel(m)

2.2 体积引入的相对标准不确定度urel(v)
主要包含加入提取溶剂V1、移取上清液的体积V2、最终乙酸乙酯复溶的体积V3。
2.2.1 加入提取溶剂引入的相对标准不确定度urel(V1)

所以移液管移取15 mL乙腈—醋酸(V乙腈∶V醋酸=99∶1)溶液引入的相对标准不确定度为:
2.2.2 移取上清液引入的相对标准不确定度urel(V2)
移取2 mL上清液使用的是2 mL A级单标线吸量管,其允许误差为±0.010 mL,参考15 mL单标线吸量管的评定过程,2 mL A级单标线吸量管引入的不确定度为:
实验室温度变化引入的标准不确定度为:
所以吸量管移取2 mL上清液引入的相对标准不确定度为:
2.2.3 乙酸乙酯复溶引入的相对标准不确定度urel(V3)
使用1 mL A级单标线吸量管移取1 mL乙酸乙酯复溶,1 mL A级单标线吸量管允许误差为±0.007 mL,参考15 mL单标线吸量管的评定过程,1 mL A级单标线吸量管引入的不确定度为:
乙酸乙酯的体积膨胀系数为1.38×10-3℃-1,则温度对乙酸乙酯体积变化引入的标准不确定度为:
所以移取1 mL乙酸乙酯复溶的相对标准不确定度为:
综上,由提取液移取、提取液净化分取和浓缩定容3个步骤引起的合成相对不确定度urel(v)为:
2.3 标准工作溶液校准过程引入的相对标准不确定度urel(c)
该相对标准不确定度主要来源:标准工作溶液的配制、标准工作曲线拟合。
2.3.1 标准工作溶液的配制引入的相对标准不确定度u1rel(c) 试验使用的水胺硫磷标样购于北京曼哈格生物科技有限公司,标准物质给出的质量浓度为1 000.0 μg/mL,其扩展不确定度为±3%(k=2),所以标准溶液的不确定度为:
10 μg/mL的标准储备液的配制:使用量程为20~200 μL的移液器移取1 000.0 μg/mL的标准原液100 μL到10 mL容量瓶,用乙酸乙酯定容至刻度。
标准溶液稀释过程:其数学模型为
(2)
式中:
cx——稀释后标准溶液的质量浓度,μg/mL;
cs——标准原液的质量浓度,μg/mL;
vy——移液器移取的体积,μL;
vr——定容时容量瓶的体积,mL。
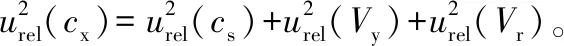
1 μg/mL储备液:用1 mL A级单标线吸管吸取10 μg/mL 的储备液1 mL到10 mL A级容量瓶中,用乙酸乙酯定容至刻度,其相对标准不确定度为:
标准工作溶液的配制:移取上述标准溶液1 mL到茶叶空白基质中,加20 μL环氧七氯B内标溶液(5 μg/mL),过膜,配制成质量浓度分别为0.005,0.01,0.05,0.1,0.5 μg/mL的标准工作溶液。1 mL标样的移取使用的是1 mL A级单标线吸量管,样品复溶时已有评定,这里不重复评定。试验选择与被测样品接近的urel(c0.01)=2.223%予以评定,则u1rel(c)=urel(c0.01)=2.223%。
2.3.2 标准工作曲线拟合引入的不确定度 5种浓度的标准工作溶液由低到高分别上机进样,得到的浓度比及面积比的数据见表2。采用仪器自带的定量软件最小二乘法拟合,得到标准工作曲线的回归方程和相关系数为:
Arf=-0.025 042+5.544 347cri,R2=0.999 997,
(3)
式中:
cri——标准溶液与内标溶液浓度比值;
Arf——标准物质峰面积与内标峰面积的比值。
为了评定茶叶中水胺硫磷残留限量时(即残留量为0.05 mg/kg)的不确定度,所以假定某阳性样品平行测定2次,得到样液中平均质量浓度为0.013 33 μg/mL,内标质量浓度为0.1 μg/mL。其与内标浓度比的相对标准不确定度按式(4)计算:
(4)

表2 线性回归方程拟合过程及结果†Table 2 Linear regression equation fitting and results
式中:
s——拟合标准工作曲线的标准偏差;
b——拟合标准工作曲线的斜率,b=5.544 347;
p——试样测定的次数,p=2;
n——标准工作溶液的测定次数,n=5;
cr——试样中水胺硫磷与内标浓度比的平均值,cr=0.133 3;

cri——标准工作溶液中水胺硫磷与内标的系列浓度比。



所以标准工作溶液校准引入的相对标准不确定度为:
2.4 重复性测量引入的不确定度urel(rep)

2.5 方法回收率引入的不确定度urel(R)
对市售茶叶按1.2试验方法进行检测,在水胺硫磷目标峰位置观察其质谱图,无法识别出水胺硫磷特征离子的样品确定为阴性样品。称取6份阴性茶叶样品,添加量为0.05 mg/kg,测定回收率,结果见表3。

表3 样品添加回收率Table 3 Results of replicate recovery tests in samples
标准不确定度采用平均值的标准偏差计算得到:

因此方法回收率引入的不确定度需进行修正。但通常情况下,检测结果满足标准要求时,不需要把回收率直接折算到检测结果中,所以将回收率以水胺硫磷纯度的方式参与到评定过程中,则
2.6 合成标准不确定度的评定
综上所述,各因素相对不确定度分别为urel(m)=2.04%,urel(v)=0.778%,urel(c)=4.35%,urel(rep)=1.88%,urel(R)=3.37%,根据不确定度传播公式对各分量进行合成:

u(ω)=0.050×6.22%=0.003 2 mg/kg。
2.7 扩展不确定度U(ω)
取包含因子k=2,置信水平P=95%,则扩展不确定度为:
U(ω)=2u(ω)=2×0.003 2=0.007 mg/kg。
测定结果:
通过QuEChERS-气相色谱—串联质谱法测定,该茶叶样品中水胺硫磷残留量结果为:
ω=(0.050±0.007) mg/kg,k=2。
3 结论
通过QuEChERS-气相色谱—串联质谱法测定茶叶中水胺硫磷残留量的不确定度分析,得出该方法的检测结果表示为ω=(0.050±0.007) mg/kg,k=2。从各标准不确定度分量数值大小可以看出,浓度c(涉及标准物质纯度、标准储备液与标准工作溶液的配制、标准工作曲线校准及内标加入量)引入的不确定度分量对合成标准不确定度贡献最大,加标回收试验引入的不确定度对合成标准不确定度的贡献次之,因此,可以通过使用不确定度更小的标样,用精度更高的量器配制标线及加内标等方式降低相关不确定度分量,也可以通过改善样品的前处理步骤(QuEChERS净化、氮吹等),减少水胺硫磷残留量的损失,特殊情况,还可以通过回收率的校准,来提高结果的准确性,保证测量结果准确、可靠。
猜你喜欢
日用电器(2022年3期)2022-04-14
中国土壤与肥料(2021年5期)2021-12-02
口腔护理用品工业(2021年4期)2021-11-02
昆明医科大学学报(2021年8期)2021-08-13
今日农业(2020年22期)2020-12-14
中国科技纵横(2019年23期)2019-02-14
读与写·下旬刊(2018年6期)2018-07-14
食品界(2017年7期)2017-08-24
科学与财富(2017年17期)2017-06-16
海峡科技与产业(2017年1期)2017-03-04
