
超高效液相色谱-串联四级杆质谱联用法测定14种蔬菜中灭蝇胺的残留
2022-11-02 01:02:52俞所银闫晴梁佳
食品与发酵工业 2022年20期
俞所银,闫晴,梁佳
1(上海市质量技术监督检验技术研究院,上海,200233)2(浙江海洋大学 食品与药学学院,浙江 舟山,316022) 3(浙江省海产品健康危害因素关键技术研究重点实验室,浙江 舟山,316021)
灭蝇胺(cyromazine)是一种用于防治多种叶菜类蔬菜、茄果类、豆类的美州斑潜蝇、豆杆黑潜蝇、葱斑潜叶蝇等的低毒杀虫剂。其对人体皮肤和眼睛具有刺激作用[1];对动物胎儿可造成骨骼发育迟缓、存活率下降等问题[2];在农作物生长过程中和动物体内都会降解代谢成三聚氰胺,经胃酸作用转化为三聚氰酸,三聚氰酸与未转化的三聚氰胺形成结晶,会导致公鼠膀胱肿瘤[3-6]。因此,国际组织和许多国家都已经制定了相应最大残留限量标准。GB/T 2763—2019《安全国家标准食品中农药最大残留限量》规定了灭蝇胺在黄瓜、豇豆、菜豆、食荚豌豆、扁豆、蚕豆、豌豆中最大残留量分别为1、0.5、0.5、0.5、0.5、0.5、0.5 mg/kg,但对韭菜、青菜、茄子、菠菜、包菜、鸡毛菜、青椒、红椒、番茄、大白菜、小青菜、芹菜等尚未明确规定。
现行有效的NY/T 1725—2009 《蔬菜中灭蝇胺残留量的测定》检测行业标准规定了黄瓜、番茄、菜豆、甘蓝、大白菜、芹菜、萝卜等蔬菜中检出限0.02 mg/kg。但日常检测发现受基质影响(韭菜样最为明显),在灭蝇胺出峰时间处有干扰峰,分离度差,且优化梯度后会有其他蔬菜基质干扰,加标样分析也往往因基质效应而掩盖,对非法添加微过量的样品造成误判,对蔬菜类质量安全监管带来困难,对百姓饮食安全和身体健康也造成了潜在的风险。相关文献报道过高效液相色谱法[7]、液相色谱-质谱法[8-11]、量子点荧光探针法[12]和超高效液相色谱-质谱联用同位素内标法[13]对灭蝇胺残留的检测,此类方法存在适用基质单一、基质影响严重、操作繁琐、保留时间过短和回收率低的不足。
故本文采用“乙腈提取/氮吹”前处理方法、基质曲线和超高效液相色谱-串联四级杆质谱联用法(ultra-high performance liquid chromatography-triple quadrupole mass spectrometry, UPLC-QQQMS)测定了韭菜、豇豆、黄瓜、青菜、茄子、菠菜、包菜、鸡毛菜、青椒、红椒、番茄、大白菜、小青菜、芹菜共14种日常送检灭蝇胺残留的新鲜蔬菜。改善了灭蝇胺在C18色谱柱上出峰早、基质效应影响判断、方法适用范围窄、操作繁琐的问题,旨在为灭蝇胺在新鲜蔬菜上的风险评估、批量快速检测上提供参考。
1 材料与方法
1.1 仪器、试剂与材料
韭菜、豇豆、黄瓜、青菜、茄子、菠菜、包菜、鸡毛菜、青椒、红椒、番茄、大白菜、小青菜、芹菜共14种新鲜蔬菜样品均为日常抽检样品。
乙腈(色谱纯),美国Fisher公司;甲酸(色谱纯),美国ACS化学试剂;乙酸铵、氯化钠(分析纯),国药集团化学试剂有限公司。灭蝇胺标准品(纯度≥99.9 %,100 μg/mL),天津阿尔塔有限公司。
6500 Q-TRAP 三重四级杆串联质谱仪,美国AB公司;Agilent 1290 超高压液相色谱仪,美国Agilent公司;5804 高速离心机,德国Eppendorf公司;SK8210LHC 型超声波清洗仪,上海科导超声仪器有限公司;G560E 涡旋振荡器,美国Scientific Industries;Milli Q 超纯水器,美国Millipore公司;KD 200 氮气吹扫仪,杭州奥盛公司;0.22 μm Filter Unit滤膜,天津博纳艾杰尔科技有限公司;分析天平(感量0.1 mg),上海梅特勒-托利多仪器公司。
1.2 标准溶液配制和标准曲线
准确移取灭蝇胺标准品,用乙腈溶解配制成质量浓度为1 μg/mL的标准储备液。用乙腈逐级稀释标准液,得到0.000 5、0.001、0.002、0.004、0.005、0.001 0、0.020、0.050和0.100 μg/mL的一系列纯溶剂标准工作溶液。
1.3 样品处理方法
按GB/T 8855抽取的新鲜蔬菜样品取可食部分切碎、混匀、密封、作为试样,表明标记,置于0~4 ℃冷藏保存待检。另一部分-20 ℃冰箱保存备样。
分别称取10 g(精确至0.000 1 g)样品于50 mL具塞玻璃管中,加入20 mL乙腈,涡旋1 min后超声提取20 min;加5 g NaCl涡旋1 min后9 500 r/min离心2 min,取5 mL上清液40 ℃氮吹干,加入1 mL乙腈溶解,0.22 μm Filter Unit滤膜过滤,UPLC-QQQMS分析。
(1)乙腈提取:分别称取10 g(精确至0.000 1 g)样品于50 mL具塞玻璃管中,加入1 μg/mL、0.4 mL灭蝇胺标准储备液(即40 ng/g),加入20 mL乙腈,涡旋1 min后超声提取20 min;加5 g NaCl涡旋1 min后9 500 r/min离心2 min,上清液0.22 μm Filter Unit滤膜过滤,UPLC-QQQMS分析。
(2)乙腈提取/氮吹:分别称取10 g(精确至0.000 1 g)样品于50 mL具塞玻璃管中,加入1 μg/mL、0.4 mL灭蝇胺标准储备液(即40 ng/g),加入20 mL乙腈,涡旋1 min后超声提取20 min;加5 g NaCl涡旋1 min后9 500 r/min离心2 min,取5 mL 上清液40 ℃氮吹干,加入1 mL乙腈溶解,0.22 μm Filter Unit滤膜过滤,UPLC-QQQMS分析。
(3)QuEChERs盐包:分别称取10 g(精确至0.000 1 g)样品于50 mL具塞玻璃管中,加入1 μg/mL、0.4 mL灭蝇胺标准储备液(即40 ng/g),加入20 mL乙腈,涡旋1 min后超声提取20 min;加入QuEChERs盐包[无水硫酸镁与无水乙酸钠的混合包(质量比4∶1,7.5 g)],涡旋1 min后9 500 r/min离心2 min,取5 mL上清液40 ℃氮吹干,加入1 mL乙腈溶解,0.22 μm Filter Unit滤膜过滤,UPLC-QQQMS分析。
(4)QuEChERs法:分别称取10 g(精确至0.000 1 g)样品于50 mL具塞玻璃管中,加入1 μg/mL、0.4 mL灭蝇胺标准储备液(即40 ng/g),加入20 mL乙腈,涡旋1 min后超声提取20 min;加入QuEChERs盐包[无水硫酸镁与无水乙酸钠的混合包(质量比4∶1,7.5 g)],涡旋1 min后9 500 r/min离心2 min,取上清液10 mL于固相萃取净化管(无水硫酸镁900 mg,N-丙基乙二胺300 mg,十八烷基硅烷键合硅胶300 mg,硅胶300 mg,石墨化碳黑90 mg)中,涡旋使充分混匀,3 000 r/min离心2 min,取上清液5 mL上清液40 ℃氮吹干,加入1 mL乙腈溶解,0.22 μm Filter Unit滤膜过滤,UPLC-QQQMS分析。
(5)SCX固相萃取柱:分别称取10 g(精确至0.000 1 g)样品于50 mL具塞玻璃管中,加入1 μg/mL、0.4 mL灭蝇胺标准储备液(即40 ng/g),加入20 mL乙腈,涡旋1 min后超声提取20 min;加5 g NaCl涡旋1 min后9 500 r/min离心2 min,取5 mL上清液40 ℃氮吹干,加入0.1 mol/L盐酸溶液定容至2 mL;转入SCX强阳离子交换萃取柱中(甲醇、水各5 mL预淋活化),2 mL 0.1 mol/L盐酸溶液润洗残渣转入SCX柱。依次水、甲醇5 mL淋洗,弃液抽干小柱,用7.5 mL氨水-甲醇(体积比5∶95)洗脱,收集洗脱液,40 ℃氮吹干,加入1 mL乙腈溶解,0.22 μm Filter Unit滤膜过滤,UPLC-QQQMS分析。
(6)CARB/SCX/PSA固相萃取柱:前处理步骤同(5)。
(7)MCX固相萃取柱:前处理步骤同(5)。
1.4 色谱条件
色谱柱:ACQUITY UPLC BEH HILIC(2.1 mm×150 mm×1.7 μm);流动相:A相:含0.1%甲酸的5 mmol/L乙酸铵溶液,B相:乙腈;等度洗脱:0~5 min,80%A;柱温为35 ℃,流速0.25 mL/min,进样体积为0.5 μL。
1.5 质谱条件
采用电喷雾离子源正离子模式(electron spray ionization, ESI+);多反应监测(multiple reaction monitoring, MRM)模式;雾化气压力0.276 MPa;辅助加热器压力0.138 MPa;气帘气压力0.172 MPa;电喷雾电压4 000 V;离子源温度350 ℃。以离子对(母离子和2个子离子)的信息比较进行定性分析,以响应值最高的子离子为定量离子,灭蝇胺在MRM模式下质谱采集参数见表1。

表1 灭蝇胺质谱参数Table 1 Chromatogram parameters of cyromazine
2 结果与分析
2.1 质谱条件优化
将质量浓度为0.1 μg/mL的灭蝇胺标准液,不接液相色谱,使用手动针泵进样方式直接连接质谱,根据灭蝇胺结构选择ESI+下全扫描,确定目标物的分子离子,然后将分子离子作为母离子,在一定的碰撞能量和碰撞气体,再次全扫描二级离子选取丰度相对较大、干扰相对较小的2个子离子分别作为定量、定性离子,并优化去簇电压、碰撞能量值,质谱参数见表1,质谱图见图1。
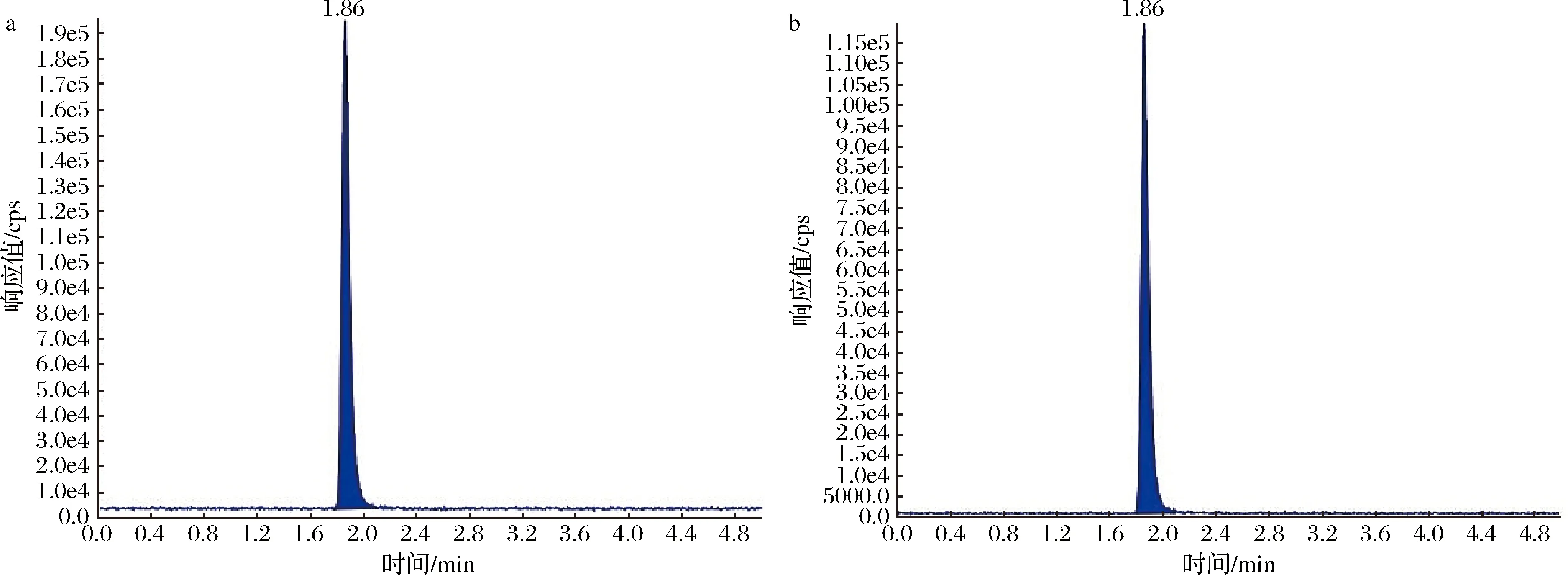
a-定量离子MYA1;b-定量离子MYA2图1 灭蝇胺标准品色谱图Fig.1 The chromatogram of cyromazine注:MYA1、MYA2均为0.1 μg/mL,乙腈配制
2.2 色谱条件优化
2.2.1 色谱柱的选择
根据灭蝇胺结构:三聚氰胺与1个三元碳环相连,正负电荷中心不重合,偶极矩非0,属于极性较大的化合物,因此在反相色谱柱上很难被保留。使用C18反相色谱柱分析发现保留时间为1.0 min左右,几乎没什么保留,可以加入离子对试剂解决,但离子对试剂难以去除且容易污染色谱柱。正相色谱柱对极性化合物有较好的保留,能得到对称的峰形,但正相色谱采用非极性流动相(如正己烷),不利于离子化,响应低不易被检测,而GB/T 20769—2008中450种农药绝大数是极性或弱极性的化合物。因此,本实验比较了Agilent SB-C18(2.1 mm×50 mm×1.7 μm)、UPLC-T3(2.1 mm×100 mm×1.8 μm)、ACQUITY UPLC BEH HILIC(2.1 mm×150 mm×1.7 μm)、氨基柱对灭蝇胺的分析。在酸性或高比例水相的流动相下(≥40%)氨基柱寿命短、因此氨基柱不适用。结果发现,在相同的质谱条件下,C18、T3柱上的灭蝇胺在0.80~0.90 min较早出峰,与有机溶剂峰或杂质峰易重合;HILIC色谱柱上灭蝇胺保留时间为2.00 min,分析时间短、峰形对称良好,见图1。因此确定了ACQUITY UPLC BEH HILIC作为分离色谱柱。
2.2.2 流动相的选择
考察乙腈-甲酸水、甲醇-甲酸水、乙腈-甲酸/乙酸铵、甲醇-甲酸/乙酸铵溶液作为流动相对灭蝇胺的色谱峰和响应值的影响。结果发现,甲酸/乙酸铵比甲酸水的响应高出1.5倍,乙腈-甲酸/乙酸铵质谱响应和峰型上优于甲醇-甲酸/乙酸铵,因而选择“乙腈-甲酸/乙酸铵”为流动相溶。进一步考察乙酸铵浓度差异,比较了含0.1%甲酸的水溶液、含0.1%甲酸的5、10、20 mmol/L乙酸铵溶液。结果发现,5、10、20 mmol/L乙酸铵溶液下灭蝇胺保留时间均在2.38 min,见图2,选择5 mmol/L低溶度的乙酸铵溶液有利于延长色谱柱、仪器使用寿命。而含0.1%甲酸的5 mmol/L乙酸铵溶液获得更好的峰形,这主要是在ESI+模式下电离时提供更多的H+,使离子化效率提高,从而提高分离效果和改善峰形。
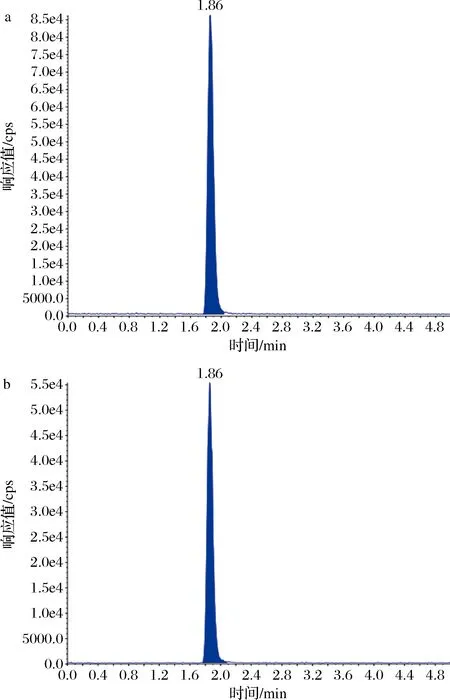
a-定量离子MYA1;b-定性离子MYA2图2 韭菜基质中加标灭蝇胺的MRM色谱图Fig.2 MRM chromatograms of cyromazine spiked in garlic chives matrix注:MYA1、MYA2均为2.0 μg/mL,韭菜基质加标样
2.2.3 洗脱梯度的选择
鉴于主要是对蔬菜中灭蝇胺残留的定量分析,因此将先前优化的8 min/单针的梯度洗脱:0.25 mL/min,0~1 min、20%A,1~3 min、20%A~80%A,3~5 min、80%A,5~5.1 min、80%A~20%A,5.1~8 min、20%A;改为6 min/单针的等度洗脱:0~5 min、80%A,缩短了分析时间且得到了较好的保留。
2.3 样品前处理方法的选择和优化
对比已报道文献中对使用过的固相萃取净化发现,石墨碳黑粉、石墨/NH2串接柱均会吸附灭蝇胺,导致回收率下降;C18、NH2固相萃取柱色素、杂质吸附效果一般,进样分析发现在目标物峰附近有杂质干扰;MCX固相萃取柱效果相对较好,回收率在65.4%~108.8%均有报道[12-13]。因此本文选择干扰较强的韭菜为基质,加标样进行比较:(1)乙腈提取、(2)乙腈/氮吹、(3)QuEChERs盐包、(4)QuEChERs法、(5)SCX、(6)CARB/SCX/PSA和(7)MCX 固相萃取柱法,结果发现,QuEChERs盐包因与干扰峰无法完全分离,导致平均加标回收率偏高;“乙腈提取/氮吹”、“MCX固相萃取柱法”平均加标回收率接近值,但“乙腈提取/氮吹”法的前处理操作更便捷;“乙腈提取/氮吹”法因浓缩后样品的化合物响应强度远远大于目标物附近的杂质响应,目标物定量更准确,并且因浓缩后样品的pH变化引起化合物峰面积更大且峰型对称,结合检测效率与成本,选择“乙腈提取/氮吹”法为最佳前处理方法,数据见表2。

表2 不同前处理法韭菜中灭蝇胺的回收率(n=3)Table 2 Recoveries of cyromazine in garlic chives by different pretreatment methods (n=3)
2.4 方法学验证
2.4.1 基质效应影响
选取14种阴性新鲜蔬菜作为空白样品,按照“1.3”方法前处理,得到14种空白基质提取液。用乙腈逐级稀释,得到0.000 5、0.001、0.002、0.005、0.020、0.050、0.100 μg/mL的一系列基质标准工作溶液。按“2.1”~“2.3”优化好的条件分析。外标法绘制基质标准曲线[其中,待测物质量浓度为横坐标(X,mg/L),对应定量离子对的峰面积为纵坐标(Y)]。样品的基质效应(matrix effect,ME)=基质标准曲线斜率/溶剂绘制的标准曲线斜率,当ME值越接近1时,表明样品的基质效应越小[14];ME<1,基质抑制作用;ME>1基质增强作用。如表3所示,14种新鲜蔬菜基质均对灭蝇胺有明显的抑制效应,且不同基质间存在明显的区别,与郝国辉等[8]、徐炜枫[14]的研究结果一致。为了减小样品的基质效应,提高方法分析的准确度,本文以空白样品提取液配制的基质标准工作曲线,外标法定量进行试验。
2.4.2 方法的线性范围、检出限和定量限
准确吸取质量浓度为1 μg/mL的灭蝇胺标准液,用14种空白基质溶液稀释成0.000 5、0.001、0.002、0.005、0.020、0.050、0.100 μg/mL的一系列基质标准工作溶液;按本试验优化的条件分析,以定量离子峰面积为纵坐标,灭蝇胺含量(μg/kg)进行线性回归。结果表明,14种新鲜蔬菜中灭蝇胺在0.000 5~0.100 μg/mL内具有良好的线性关系,以信噪比S/N=3、S/N=10分别计算出检出限、定量限,见表3。

表3 14种新鲜蔬菜中灭蝇胺的基质效应和线性方程、检出限和定量限Table 3 Matrix effects, linear equations, limits of detection and limits of quantification of cyromazine in 14 kinds of fresh vegetables
2.4.3 方法的准确度和精密度
称取14种(n=3)对应阴性蔬菜样品,按GB/T 27404—2008《实验室质量控制规范食品理化检测》[15]的方法技术要求,分别添加2.0、4.0、20.0 μg/kg(定量限的1倍、2倍和10倍)[16],按“1.3”方法前处理,连续测定6次,结果表明,方法的回收率80%~113%,精密度0.56%~7.21%,见表4。
2.5 实际样品测定
采用已建立的方法对上海市、扬州市、苏州市姑苏区抽查新鲜蔬菜60份进行灭蝇胺检测,其中38份样品被检测出,残留量为0.8~40 μg/kg,其余均未被检出;而对比现行有效NY/T 1725—2009《蔬菜中灭蝇胺残留量的测定》标准,样品中均未检出灭蝇胺,且基质曲线回归方程也无法测出,这主要由仪器灵敏度差异决定。6500 Q-TRAP ESI+扫描、MRM模式下1pg利血平,信噪比≥160 000∶1,50 fg和1 pg 利血平分别连续进样10次,峰面积变异系数<2%,灵敏度高于液相色谱法数量级以上,因此优点样品前处理更简便、抗干扰强,实验表明UPLC-QQQMS在测定蔬菜中灭蝇胺低含量残留方法方面具有明显优势。

表4 14种新鲜蔬菜中灭蝇胺残留量的回收率 和精密度(n=6)Table 4 Recoveries and relative standard deviations (RSDs) of cyromazine in 14 kinds of fresh vegetables(n=6)
3 结论
本试验通过“乙腈提取/氮吹”前处理方法、基质曲线结合UPLC-QQQMS测定了韭菜、豇豆、黄瓜、青菜、茄子、菠菜、包菜、鸡毛菜、青椒、红椒、番茄、大白菜、小青菜、芹菜共14种日常送检灭蝇胺残留的新鲜蔬菜。该方法前处理简单、操作便捷、检测快速,方法重现性好、准确度高且精密度好,同时解决了现行NY/T 1725—2009 《蔬菜中灭蝇胺残留量的测定》行业标准中基质影响造成HPLC分析方法的缺陷;为灭蝇胺在蔬菜上的风险评估、批量快速检测提供参考。
猜你喜欢
北京航空航天大学学报(2022年7期)2022-08-06 07:28:48
食品安全导刊(2021年20期)2021-08-30 06:39:44
食品安全导刊(2020年21期)2020-09-07 09:14:04
农家科技中旬版(2019年9期)2019-10-08 05:27:47
山西农业科学(2019年6期)2019-06-19 07:14:40
中央民族大学学报(自然科学版)(2018年1期)2018-06-27 01:27:48
农村百事通(2017年14期)2017-09-21 17:03:08
山东工业技术(2016年13期)2016-06-29 09:05:13
工业设计(2016年10期)2016-04-16 02:43:56
制造技术与机床(2015年10期)2015-04-09 07:06:04
