
底物亲和设计提高腈水解酶Nit6803活性
2022-11-02 01:29刘欣悦韩来闯刘中美
食品与发酵工业 2022年20期
刘欣悦,韩来闯,刘中美
(江南大学 生物工程学院,江苏 无锡,214122)
腈水解酶(Nitrilase,EC 3.5.5.1),是一类可以将氰基一步水解为羧酸的酶,广泛分布于植物、动物、细菌、真菌和古菌中[1-3]。除了天然酶资源丰富、反应条件温和以外,Nitrilase还具有较强的区域选择性和立体选择性,因此是多种重要大宗化工品、药物中间体的理想生物催化剂[4]。然而,目前鉴定的天然Nitrilase底物谱较窄、活性低、热稳定性差,极大地限制了其规模应用[5]。近些年,人们不断尝试通过天然菌种分离、宏基因组分析等手段挖掘新型Nitrilase[6]。但是受制于酶活性与稳定性间的“trade-off”效应,嗜温生物来源的Nitrilase活性较好但稳定性差;嗜热生物的Nitrilase稳定性强但活性极低[7-8]。因此,直接挖掘得到高性能天然Nitrilase的希望十分渺茫。
酶分子改造是提升酶学性质的有效手段,主要包含3种基本策略:定向进化、理性设计及半理性设计,针对Nitrilase的分子改造也获得了一定效果[9]。例如,SCHREINER等[10]基于易错PCR对来源于AlcaligenesfaecalisJM3的Nitrilase进行定向进化,提高其活性及对低pH的耐受性。定向进化最为依赖高通量筛选方法,虽然一些显色试剂和pH指示剂已被用来筛选Nitrilase的活性,但是依然远未达到理想的筛选通量[11]。因此,现阶段构建大容量的突变体库对Nitrilase定向进化依然十分困难。而另一方面,随着蛋白质结构预测、功能分析软件不断开发,以及计算机算力的指数增长,蛋白质半理性、理性设计的易用性和可靠性快速提升。例如,基于人工智能的蛋白质全原子结构预测平台AlphaFold2的开源,将结构生物学从实验解析带入了大规模预测的新阶段[12]。华盛顿大学David Baker课题组开发的Rosetta套件功能多样,且具备良好的可编程性,经过多年发展,已经成为蛋白质分子改造、人工蛋白质设计的强大工具[13]。而分子对接、分子动力学模拟及量子化学计算的广泛应用,为在微观上观察酶分子运动、解析催化机制提供了可能[14-16]。多种理性设计策略在Nitrilase的改造上也得到成功应用,大幅提升其酶学性质。例如,汤晓芒[17]对来源于古菌Pyrococcusabyssi的腈水解酶PaNit通过分子模拟及MaCrodox程序和TK-SA模型等计算方法获得热稳定性增强且仍具备活力的突变体。YU等[18]通过催化口袋底物结合位点的重设计实现了腈水解酶对映选择性的调控,为腈水解酶在不对称催化中的应用奠定基础。对杂合腈水解酶BaNIT进行组合突变设计,可提升其可溶表达、对映选择性及活性[19]。
众所周知,酶与底物的结合是其发挥催化功能的先决条件。大量研究表明,优化催化口袋,提升酶与底物的亲和力,能够有效提高酶活力[20]。基于此,本研究提出一种酶-底物亲和设计策略,以提高酶活力。如图1所示,首先,设计催化口袋残基的单点饱和突变,并用Cartesian_ddG方法过滤对蛋白质稳定性不利影响的突变。然后使用自由能微扰(free energy perturbation,FEP)方法计算底物亲和力变化,经酶活力表征获得正向突变。最后,进一步做组合突变设计,提升活性。本研究选择来源于Syechocystissp.PCC6803的腈水解酶Nit6803作为研究对象,该酶底物谱较广,但酶活力较低,无法满足应用需求[21-22]。经过本研究提出的底物亲和策略改造,获得了催化烟腈活性提升3.56倍的突变体,证明该策略的有效性。

图1 酶-底物亲和改造策略Fig.1 Enzyme-substrate affinity modification strategy
1 材料与方法
1.1 实验材料
1.1.1 菌种与质粒
克隆宿主EscherichiacoilJM109、表达宿主EscherichiacoliER2566、表达质粒pET-24a(+),本实验室保藏。
1.1.2 主要试剂
胰蛋白胨、酵母提取物,Oxoid公司;氯化钠、氯化钾、磷酸氢二钠、磷酸二氢钾、磷酸二氢钠、咪唑、烟酸,国药有限化学试剂有限公司;3-氰基吡啶,Sigma公司;异丙基硫代半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG)、卡那霉素,上海生工生物工程有限公司;培养基中的卡那霉素终质量浓度为50 μg/mL。
1.1.3 培养基
LB培养基(g/L):胰蛋白胨10,酵母提取物5,氯化钠10。固体培养基加入20 g/L琼脂粉。2×YT培养基(g/L):胰蛋白胨16,酵母提取物10,氯化钠5。
1.1.4 缓冲液
PBS缓冲液:氯化钠8 g,氯化钾0.2 g,磷酸氢二钠1.42 g,磷酸二氢钾0.27 g,调pH至7.4,用去离子水定容至1 L。Binding buffer:0.2 mol/L 磷酸二氢钠,0.2 mol/L 磷酸氢二钠,调pH为7.4,加入20 mmol/L 咪唑。Washing buffer:上述Binding buffer中咪唑浓度调整至500 mmol/L。
1.2 实验方法
1.2.1 分子对接
从PDB数据库获得Nit6803的三维结构(PDB code:3 WUY),从PubChem数据库(https://pubchem.ncbi.nlm.nih.gov/)获得底物3-氰基吡啶的三维结构。使用软件Schrödinger的Glide模块进行分子对接(extra precision模式),生成50个poses结果。结合对接打分及视觉判断,选择合适的对接pose,获得Nit6803结合3-氰基吡啶的复合体结构。定义距离底物5 Å以内的氨基酸为组成催化口袋的残基。使用在线工具PLIP(https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index)分析催化口袋残基与底物间的相互作用。
1.2.2 折叠自由能计算
使用基于Rosetta的Cartesian_ddG方法计算点突变对蛋白质折叠自由能的影响[23]。首先,通过内坐标relax对Nit6803的初始晶体结构优化,优化时对蛋白骨架及残基侧链重原子添加坐标限制势。选择能量最低的结构进行笛卡尔坐标relax,进一步优化结构。笛卡尔坐标relax过程中不限制蛋白骨架及残基侧链重原子坐标。使用两步relax优化后的结构进行Cartesian_ddG计算,根据相比于野生型的G评价突变对酶稳定性的影响。定义G≤20.93 kJ/mol为对蛋白稳定性没有显著影响。
1.2.3 酶-底物结合自由能计算
使用自由能微扰/汉密尔顿副本交换分子动力学(FEP/hamiltonian replica exchange molecular dynamics, FEP/HREMD)计算酶与底物的结合自由能。计算所需文件使用在线工具CHARMM-GUI(https://charmm-gui.org/)生成,然后使用NAMD 2.14软件进行计算,副本数量设定为32。计算体系为边界10 Å的正方体盒子,其中填充含有0.15 mol/L K+和Cl-的中性TIP3显性水模型。
1.2.4 分子动力学模拟
使用软件NAMD 2.14进行CHARMM36力场的分子动力学模拟计算。3-氰基吡啶的拓扑文件及力场参数由CGenFF生成。计算体系为边界10 Å的正方体盒子,其中填充含有0.15 mol/L K+和Cl-的中性TIP3显性水模型。计算步长为2 fs,非键相互作用的cutoff设定为12 Å。使用Particle Mesh Ewald(PME)实现周期性边界条件的应用。动力学模拟过程包含1 ns在NVT系综下的水平衡,60 ps NVT系综下0~300 K的升温,以及20 ns在NPT系统下300 K的常规动力学模拟。使用Bio3D分析模拟过程的均方根偏差(root mean square deviation, RMSD)及不同残基骨架的均方根波动(root mean square fluctuation, RMSF)。
1.2.5 定点突变
腈水解酶Nit6803基因,由苏州金唯智公司进行密码子优化和基因合成,并连接至表达载体pET-24a(+)NdeⅠ和XhoⅠ酶切位点之间,6×His-tag融合在Nit6803的C端,得到表达质粒pET24a-Nit6803。以该质粒作为模板,通过全质粒PCR及无缝克隆构建Nit6803突变体表达质粒。50 μL的PCR扩增体系包含:2×PrimeSTARTMMax DNA Polymerase(Takara,日本)25 μL、上下游引物各2 μL,模板0.5 μL、ddH2O 20.5 μL。PCR扩增反应条件为98 ℃预变性3 min,30个循环的98 ℃变性15 s、55 ℃退火30 s、72 ℃延伸1 min 45 s,最后72 ℃延伸5 min。PCR产物用DpnⅠ消化酶消化2~3 h,以去除模板。纯化后的片段使用ABclonal MultiF Seamless Assembly Mix(ABclonal Technology,中国)通过引物的同源臂序列无缝克隆自组装,进而转化至E.coliJM109感受态细胞中,涂布于LB平板,37 ℃过夜培养。接取单菌落培养后,送苏州金唯智公司测序验证。本研究所用引物见表1。
1.2.6 酶的表达与纯化
将表达质粒转化至E.coliER2566感受态细胞,单菌落接种至含有3 mL LB的试管中,37 ℃、200 r/min培养8~10 h。以2%(体积分数)的接种量转接种子液到含有5 mL LB试管中,37 ℃、200 r/min培养至OD600≈0.6,添加终浓度为0.5 mmo/L的IPTG。在25 ℃、200 r/min下继续培养12~16 h,收取菌液用于全细胞酶活力检测。

表1 本文所用引物及其序列Table 1 Primers used in this study
挑取单菌落至含有3 mL LB的试管中,37 ℃、200 r/min条件下培养8~10 h。以1%(体积分数)的接种量转接种子液至含有100 mL 2×YT培养基的三角瓶中,37 ℃、200 r/min条件下培养至OD600≈0.6,加入终浓度为0.5 mmol/L的IPTG,温度调整至25 ℃继续培养12~16 h。经10 000 r/min离心3 min收集菌体,弃上清液,用20 mL Binding buffer重悬,于冰水混合物中超声破碎。破碎液在4 ℃、12 000 r/min条件下离心30 min,取上清液过0.22 μm滤膜。采用亲和层析的方法纯化腈水解酶,纯化柱为GE公司的HisTrap HP 5 mL柱。用Binding buffer平衡纯化柱后上样,然后用5~10个柱体积的Binding buffer洗去杂蛋白,目的蛋白用Washing buffer梯度洗脱并收集活性蛋白。蛋白浓度使用Bradford法定量,SDS-PAGE检测蛋白纯度。
1.2.7 酶活力测定
全细胞活性测定:使用PBS将细胞OD600调至2。取100 μL菌液,加入400 μL PBS和500 μL 100 mmol/L 3-氰基吡啶溶液,充分混匀后于37 ℃金属浴中反应10 min。反应液12 000 r/min离心10 min,取上清液经0.22 μm滤膜过滤。纯酶活性测定:用PBS将纯酶的质量浓度稀释至0.5 mg/mL,取10 μL至1.5 mL离心管中,加入490 μL 100 mmol/L 3-氰基吡啶溶液,充分混匀后于37 ℃金属浴中反应10 min。加入500 μL乙腈终止反应,过0.22 μm滤膜。
使用HPLC测定反应产物中烟酸含量。色谱柱为Diamonsil C18(2) 5 μm 250 mm×4.6 mm(迪马科技,中国),流动相为乙腈和水的混合液[V(水)∶V(乙腈)=2∶1]。检测波长为210 mm,柱温40 ℃,流动相流速为0.6 mL/min,进样量10 μL,检测间隔为10 min,每组数据至少进行3次平行实验。单位酶活力定义为在37 ℃下每分钟催化3-氰基吡啶生成1 μmol 烟酸所需要的酶量(U)。比酶活力为每毫克腈水解酶所具有的酶活力(U/mg)。
1.2.8 热稳定性测定
将酶质量浓度调整至0.5 mg/mL,在金属浴40 ℃和50 ℃下分别处理0、0.5、1、2、4 h取样,测定其酶活力,以未经热处理的酶活力作为100%,分析其稳定性。
1.2.9 分批补加3-氰基吡啶催化合成烟酸
反应体系为50 mL于37 ℃、200 r/min的摇床中进行催化合成,其中菌液OD600为10,每次添加50 mmol/L 3-氰基吡啶,共添加10次至终浓度为500 mmol/L。在一定时间取样利用HPLC检测烟酸生成量与底物残留量。
2 结果与分析
2.1 单点突变设计及表征
利用软件Schrödinger将3-氰基吡啶对接至Nit6803的晶体结构中,获得酶-底物复合体结构(图2-a)。定义距底物5 Å范围内的氨基酸残基组成催化口袋,包含10个位点:Glu53、Tyr59、Phe64、Thr139、Tyr140、His141、Trp170、Met197、Val198和Phe202(图2-b)。由于Pro对蛋白质的二级结构影响较大,因此不考虑突变为Pro的设计。使用Rosetta的Cartesian_G计算催化口袋180个单点突变的折叠自由能,并计算相比于野生型(wild type, WT)的变化(G),计算结果如图2-c所示。定义G≤5为对蛋白的稳定性影响不大,共包含86个单点突变。进一步利用FEP/HREMD计算这些突变体的底物结合自由能,并与WT相比较(Gbinding)。定义Gbinding≤-1为对底物亲和有显著影响,包含10个单点突变体,结果见图2-d。
构建10个设计的单点突变体,诱导表达后测定全细胞酶活力。以WT的酶活力作为100%,结果如图2-e所示,突变体F64Y、W170G的酶活力显著高于野生型,分别达到野生型的200%、150%,V198D与野生型酶活力接近,其余突变体酶活力明显降低。进一步,对F64Y和W170G纯化并测定纯酶比酶活力,分别为(10.04±0.24)U/mg和(7.1±0.41)U/mg,显著高于WT(4.93±0.48)U/mg(图2-f)。由于催化口袋残基较多,其组合突变的数量庞大,难以穷举设计,因此首先对单点饱和突变进行设计和表征。以上结果表明,通过底物亲和力设计策略,成功获得活性显著提升的单点突变体,证明了策略的有效性。

a-酶-底物复合体结构;b-催化口袋示意图;c-单点突变Cartesian_G折叠自由能变化;d-单点突变FEP结合自由能变化; e-单点突变体全细胞相对酶活力;f-单点突变体比酶活力图2 单点突变设计及酶活力表征Fig.2 Single-point mutation design and enzyme activity characterization
2.2 组合突变设计及表征
由单点突变体的酶活力结果可知Phe64和Trp170两个位点对于该酶催化活性有显著影响,因此基于这两个位点做组合突变设计。与单点突变的设计策略类似,首先使用Cartesian_G计算324个组合突变体的折叠自由能,并计算相比于WT的变化(G)。计算结果如图3-a所示,G≤5的有37个组合突变,且集中于F64H、F64 W、F64Y和W170F、W170Y几种突变情况。这说明,由于WT中Phe64和Trp170都是大位阻芳香侧链残基,突变成其他类型侧列氨基酸后对整体的折叠自由能影响较大。进一步,利用FEP/HREMD对这些突变体的底物结合自由能进行计算,结果显示,共有14个突变体的底物结合自由能相比于WT降低(图3-b)。其中,仅有F64Y/W170G组合突变的底物结合自由能比单点突变的F64Y更低。
选择Gbinding最低的F64Y/W170G,测定全细胞活性,结果显示,F64Y/W170G的全细胞活性为野生型的476%(图3-c)。对F64Y/W170G纯酶的酶活力进行测定,其比酶活力为(22.48±0.64) U/mg,为野生型的4.56倍(图3-d)。该结果表明,针对Phe64和Trp170两个位点进行组合突变设计,并通过底物亲和计算,得到相比于单点突变进一步提升活性的突变,证明了本文提出的设计策略的可靠性。

a-组合突变Cartesian_ΔΔG折叠自由能变化;b-组合突变FEP结合自由能变化;c-组合突变全细胞相对酶活力;d-组合突变比酶活力图3 组合突变设计及酶活力表征Fig.3 Combinatorial mutation design and enzyme activity characterization
为了解析组合突变F64Y/W170G显著提升Nit6803催化3-氰基吡啶活性的分子机制,对WT和F64Y/W170G与底物结合复合体做20 ns的分子动力学模拟计算(图4-a)。由于活性中心与待催化基团的距离与酶的活性高低关系密切[24],我们分析了模拟过程中Nit6803活性中心Cys169的巯基和底物3-氰基吡啶的氰基之间的距离。结果表明,得益于F64Y/W170G与底物更高的亲和力,其活性中心与底物间的距离相比WT更近(图4-b),这也是其具备更高活性的重要原因。进一步,对WT和F64Y/W170G与底物间的相互作用分析,结果表明,F64Y/W170G保持了和WT类似的酶-底物相互作用:Tyr140与3-氰基吡啶的吡啶环形成π-π堆叠,Phe64、Val198与3-氰基吡啶存在疏水相互作用(图4-c)。
2.3 突变体的热稳定性分析
本研究提出的设计策略首先使用Cartesian_G排除折叠自由能显著提升的突变,以防止突变对热稳定性的不利影响。为了验证该步骤的有效性,测定WT与F64Y/W170G分别在40、50 ℃下处理后的残余酶活力。以未热处理的酶活力作为100%,结果如图5-a所示,F64Y/W170G与WT的稳定性基本一致,甚至在40 ℃下F64Y/W170G稳定性略优于WT。对未结合底物的WT和突变体进行动力学模拟分析,结果如图5-b~图5-d所示,RMSD结果显示突变体的整体波动小于野生型。F64Y/W170G的RMSF和回转半径Rg与WT相比没有明显差异,进一步证明了F64Y/W170G和WT相比,不论是各残基还是蛋白整体的稳定性差别不大。以上结果表明通过本研究提出的设计策略,成功大幅提高了Nit6803活性的同时没有影响其热稳定性。
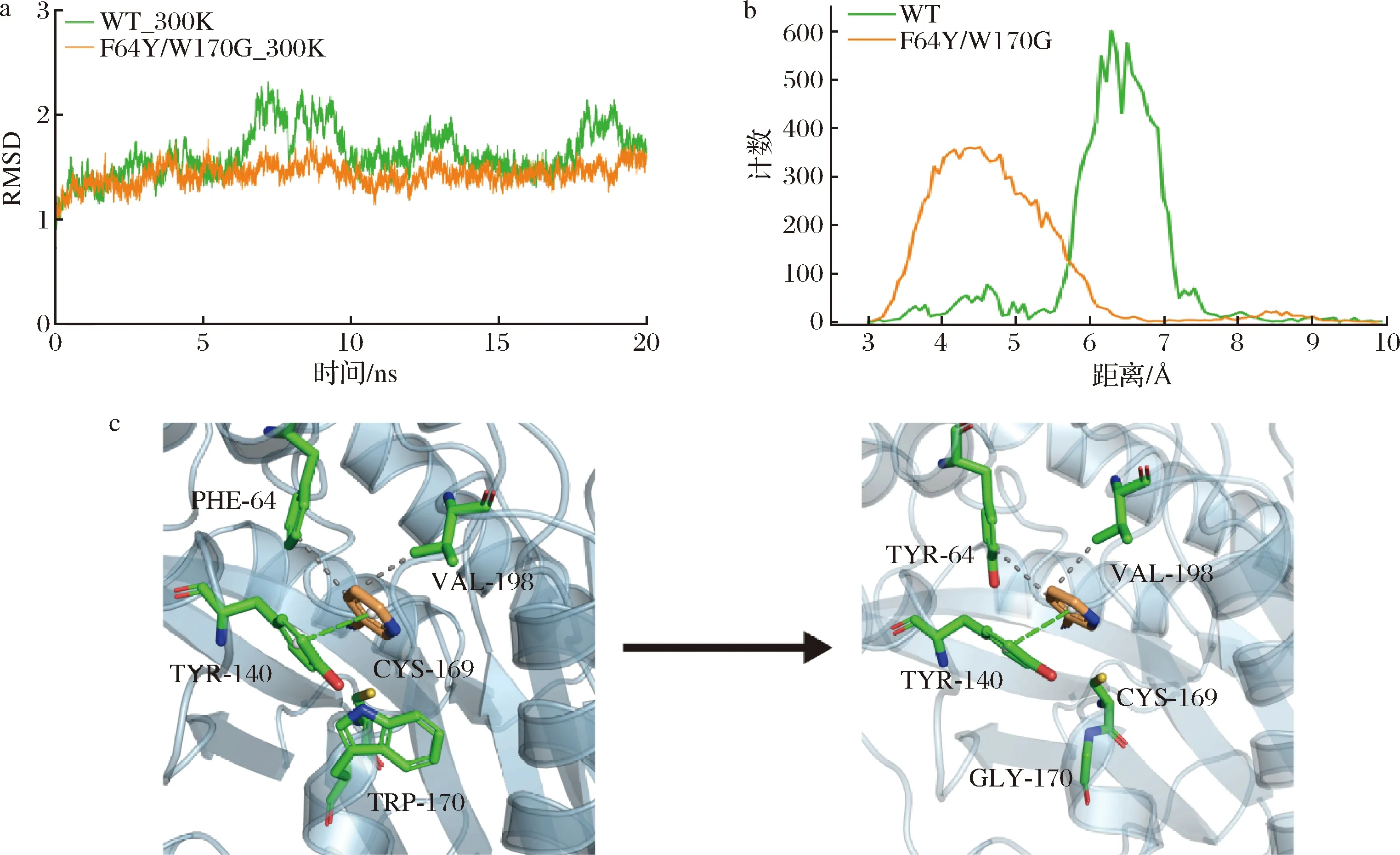
a-300 K下野生型和突变体F64Y/W170G的RMSD值;b-野生型和突变体的活性中心Cys169巯基与底物氰基之间的距离; c-野生型和突变体F64Y/W170G与底物间的相互作用图4 突变体F64Y/W170G分子动力学模拟分析及与底物之间相互作用Fig.4 Molecular dynamics simulation analysis and interaction with substrate of mutant F64Y/W170G

a-野生型与突变体的热稳定性;b-400 K下野生型与突变体的RMSD值;c-400 K下野生型与突变体的RMSF值; d-400 K下野生型与突变体的回转半径Rg值图5 突变体F64Y/W170G的热稳定性分析Fig.5 Thermal stability analysis of mutant F64Y/W170G
2.4 分批补加3-氰基吡啶催化合成烟酸
首先添加50 mmol/L底物进行全细胞催化反应,观察在不同时间的底物残留和产物生成情况,如图6-a所示,WT在40 min可以转化90%的3-氰基吡啶,在60 min完全转化;F64Y/W170G在20 min可以转化90%的底物,25 min可完全转化为产物。说明F64Y/W170G的催化速率更快,同时确定WT与F64Y/W170G分批补加底物的时间间隔分别为40、20 min。因为采用的等OD值的菌液进行催化,为了减少表达量差异对于催化能力的影响,通过SDS-PAGE检测两者的表达情况,结果如图6-b所示,WT与F64Y/W170G相同OD值下表达基本一致。说明突变体确实是由于酶活力的提升导致催化速率的加快。通过分批补加底物进行全细胞催化,F64Y/W170G结果如图6-c所示,转化95%的底物生成烟酸需要250 min,在430 min时检测不到底物,说明无底物残留,完全转化为烟酸。WT结果如图6-d所示,转化95%的底物需要540 min且到600 min时仍可检测到底物存在。综上所述,F64Y/W170G催化3-氰基吡啶能力强于WT,分批补加共500 mmol/L 3-氰基吡啶后达到95%的转化率WT需要9 h,而F64Y/W170G只需4 h,相对于野生型来大幅缩短了催化时间。

M-Marker,1-WT,2-F64Y/W170G a-野生型与突变体对于50 mmol/L 3-氰基吡啶的反应过程;b-野生型与突变体蛋白表达情况 c-F64Y/W170G分批补加3-氰基吡啶催化结果;d-WT分批补加3-氰基吡啶催化结果图6 分批补加3-氰基吡啶催化合成烟酸Fig.6 Catalytic synthesis of nicotinic acid by adding 3-cyanopyridine in batches
3 结论
本文提出了一种结合Cartesian_G和FEP/HREMD的酶-底物亲和力设计策略,并利用该策略成功提升了腈水解酶Nit6803对3-氰基吡啶的催化活性,同时没有对酶的热稳定性造成不利影响。众所周知,酶活性与稳定性之间的“trade-off”效应一直是酶分子改造的瓶颈,而本研究提出的策略为突破这一瓶颈的理性设计提供了新的范例。另外,本研究提出的策略直接计算所有单点突变和组合突变的影响,相比于传统的基于丙氨酸扫描的半理性设计方法,具有更大的覆盖范围。
天然腈水解酶活性较低,严重限制了其工业应用。本研究通过理性设计和分子改造,Nit6803催化3-氰基吡啶的活性显著提升,其中组合突变F64Y/W170G的比酶活力达到了WT的4.56倍。通过分批补加底物进行全细胞催化时,催化500 mmol/L 3-氰基吡啶使达到95%的转化率野生型需要9 h,突变体只需要4 h,大幅缩短了催化时间,有望应用于工业生产。而本研究提出的设计策略可以成为酶分子催化不同反应的通用改造方法,不仅推进腈水解酶的应用,也为其他酶的改造提供了新的思路。
猜你喜欢
汽车实用技术(2022年11期)2022-06-20
科学导报(2022年21期)2022-04-10
导航定位学报(2022年1期)2022-02-17
现代农药(2022年1期)2022-02-15
船海工程(2021年3期)2021-06-28
教育周报·教育论坛(2020年3期)2020-10-21
科技资讯(2018年16期)2018-10-26
科技信息·下旬刊(2018年8期)2018-10-21
作文与考试·小学高年级版(2017年16期)2017-08-14
科技资讯(2017年12期)2017-06-09
