
基于乙基纤维素颗粒的亚麻籽油微胶囊载体的构建与表征
2022-10-29 06:07张若宁刘楠张彦慧杨静怡高彦祥毛立科
食品研究与开发 2022年20期
张若宁,刘楠,张彦慧,杨静怡,高彦祥,毛立科
(中国农业大学食品科学与营养工程学院,北京 100083)
微胶囊技术一般指由天然或合成的高分子材料作为壁材,包埋液体、固体或气体(芯材)形成微胶囊的技术。包埋的主要目的是保护不稳定的芯材,提高其贮藏稳定性,并控制芯材的释放[1]。微胶囊技术可以保护功能性油脂,减少其受空气、光照、水分等外界环境因素的影响[2]。此外,油脂经微胶囊化后形成粉末,应用范围不断拓宽。目前微胶囊技术众多,例如界面聚合法、挤压法、喷雾干燥法、反溶剂法等[1],其中喷雾干燥法使用最为广泛。但喷雾干燥法需要使用体积庞大、价格昂贵的设备,并且具有设备整体热效率低、产品需要进一步加工(如二次造粒)等缺点,其应用范围存在局限性。
反溶剂法操作方法简单,制备条件温和,能耗低[3],因此受到广泛关注。该方法基于聚合物在不同溶剂中的溶解性差异,通过降低聚合物在溶剂中的溶解度,在油滴表面形成颗粒并固化成壁材,直接形成微胶囊。有研究通过反溶剂法构建载油的玉米醇溶蛋白微胶囊,发现油脂稳定性良好,但制备过程中需要旋转蒸发去除乙醇,工序较为复杂[4]。此外,蛋白质基壁材的性质易受到高温和酸碱性环境的影响,在实际应用中存在一定局限性。纤维素基壁材因其来源广泛,环境稳定性优良,而受到广泛关注。乙基纤维素(ethyl cellulose,EC)是一种纤维素衍生物,具有生物相容性高、可降解、成膜性好等优点。EC通过反溶剂法形成颗粒,可以包埋生育酚[5]、杨梅多酚[6]等功能性成分,但利用EC颗粒包埋功能性油脂的相关研究较少。亚麻籽油(linseed oil,LO)含有丰富的 ω-3脂肪酸,包埋亚麻籽油可以有效减少其与环境的接触,减少油脂氧化和调控油脂的消化[7]。各种添加剂可以影响壁材-芯材的相互作用,以及界面组成及结构,并进而影响微胶囊的理化性质(大小、形状和稳定性)[8-9],但在EC微胶囊体系中添加剂的作用并不明确。
本文选用乙基纤维素,以反溶剂法制备颗粒的同时包埋富含ω-3脂肪酸的亚麻籽油,研究大分子胶体明胶(gelatin,G)、小分子乳化剂吐温80(Tween 80,TW)和司盘 80(Span 80,SP)这 3 种添加剂对 LO/EC微胶囊理化性质及LO稳定性和释放特性的影响规律,以期提高LO/EC微胶囊的应用品质。
1 材料与方法
1.1 材料与试剂
乙基纤维素(黏度 9 mPa·s~11 mPa·s,5%甲苯/乙醇,48%乙氧基):上海阿拉丁试剂有限公司;亚麻籽油:内蒙古格琳诺尔生物有限公司;吐温80(食品级)、无水乙醇(>99%)、石油醚、甲醇、氯仿(分析纯):北京化工厂;明胶(食品级):河北格贝达生物科技有限公司;司盘80(食品级):山东优锁化工科技有限公司;黏蛋白、胆盐(B3301)、胰液素(P1750)、胰脂肪酶(酶活100U/mg)、胃蛋白酶(酶活400U/mg):美国Sigma公司。
1.2 仪器与设备
S22-2型恒温磁力搅拌器:上海司乐仪器有限公司;MVS-1型漩涡混合器:北京金北德工贸有限公司;AR1140型分析天平:上海奥豪斯国际贸易有限公司;BT301F蠕动泵:河北保定雷弗有限公司;Spectrum100傅里叶变换红外分光光度计:美国Perkin-Elmer公司;DSC-60差示扫描量热仪:日本岛津公司;Ultraturax T25高速剪切机:德国IKA公司;LS 13 230激光颗粒粒度分析仪:美国贝克曼库尔特公司;UV-1800紫外可见分光光度计:日本Shimadzu公司;SU8010场发射扫描电子显微镜:日本日立公司;Orion Star型pH计:美国Thermo Scientific公司。
1.3 方法
1.3.1 LO/EC微胶囊的制备
1)参考Filippidi等[4]的方法,并有所改进。将EC粉末溶解在90%乙醇中,制备5%的EC-乙醇溶液。将LO与EC-乙醇溶液在4 600 r/min下混合搅拌30 s,其中LO与EC的质量比为2∶1。使用蠕动泵以2 mL/min的速率加入水,保持4 600 r/min转速和40℃水浴使EC沉积,水与混合溶液的质量比为1∶1。室温静置,以4 200 r/min离心5 min,收集下层沉淀,沉淀用水洗涤3次。将微胶囊平铺于表面皿中,25℃干燥24 h至其水分含量<0.8%,得到的微胶囊简称为EC-P。
2)在步骤1)的基础上,控制其他条件不变,改变乳化剂种类(G、TW、SP)。以总溶液质量为基准,乳化剂质量分数均为0.5%,其中G、TW加入水中溶解,SP加入亚麻籽油中溶解。所制备的微胶囊分别简称为EC-G、EC-TW、EC-SP。)
1.3.2 LO/EC微胶囊性质的表征
1.3.2.1 粒径测定
参考文献[10]的方法,将0.05 g LO/EC微胶囊分散在50 mL去离子水中,磁力搅拌样品约10 min后,取适量的LO/EC微胶囊分散液,使用激光颗粒粒度分析仪测定其粒径大小及粒径分布。设定分散相和连续相的相对折射率分别为1.47和1.3。
1.3.2.2 微观结构观察
将干燥后的样品用导电胶粘至不锈钢样品台上,对其进行真空镀金,采用场发射扫描电子显微镜在加速电压3.0 kV下观察样品的微观形貌。
1.3.2.3 LO/EC微胶囊的表面油含量、总油含量、包埋率计算
称量微胶囊样品约1 g(精确到0.001 g,记为m1)放置在滤纸包(质量记为m2)中。滤纸包加入石油醚浸泡,轻微摇晃1 min。待石油醚在通风橱挥发2 h后,将滤纸包放入50℃的烘箱中烘干至恒重(m3),计算表面油含量,公式如下。
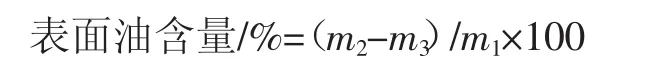
称取一定质量(m4)的微胶囊样品于滤纸包(质量记为m5)中,加入20 mL石油醚,超声15 min。待石油醚在通风橱挥发2 h后,将滤纸包置于50℃烘箱中烘干至恒重(m6)。计算总油含量和包埋率,公式如下。

1.3.2.4 傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FTIR)分析
采用傅里叶变换红外分光光度计,通过溴化钾压片法对样品进行红外光谱扫描[11]。称取2 mg样品与198 mg溴化钾粉末混合,用玛瑙研钵磨成均匀粉末,进行压片。扫描光谱范围设置为4000 cm-1~500 cm-1,扫描次数为64次,分辨率为4cm-1。以溴化钾的压片作为空白对照。对光谱曲线进行平滑处理和基线自动校正。
1.3.2.5 差示扫描量热(differential scanning calorimetry,DSC)分析
称取3 mg~5 mg左右的样品置于铝制样品盒中,密封,铝盒中央扎孔,以密封的空铝盒作为空白对照,温度范围为50℃~250℃,升温速率为10℃/min,干燥氮气的流动速度为50 mL/min。用软件TA-60WS对热曲线进行分析。
1.3.3 贮藏稳定性分析
采用过氧化值描述油脂的氧化稳定性[12]。亚麻籽油和LO/EC微胶囊在黑暗中于37℃贮藏12 d,每隔3 d取样测定。将一定量的样品溶解在5 mL氯仿/甲醇(7∶3,体积比)混合物中,涡旋 2 s~4 s,超声 10 min。然后向混合物中加入25 μL硫氰酸钾溶液(0.3 g/mL)和25 μL 氯化亚铁溶液(0.003 5 g/mL),涡旋 2 s~4 s,反应5 min。以不加样品组为空白对照,使用紫外可见分光光度计在500 nm处测定吸光度。以Fe3+标准溶液测定过氧化值标准曲线。结果以meq/kg的形式表示。
1.3.4 模拟体外消化
模拟体外消化试验过程中水浴温度保持在37℃,振荡速度为100 r/min。模拟唾液(artificial saliva,AS)、模拟胃液(simulated gastric fluid,SGF)和模拟肠液(simulated intestinal fluid,SIF)3种消化液的主要成分组成如表1所示[13]。
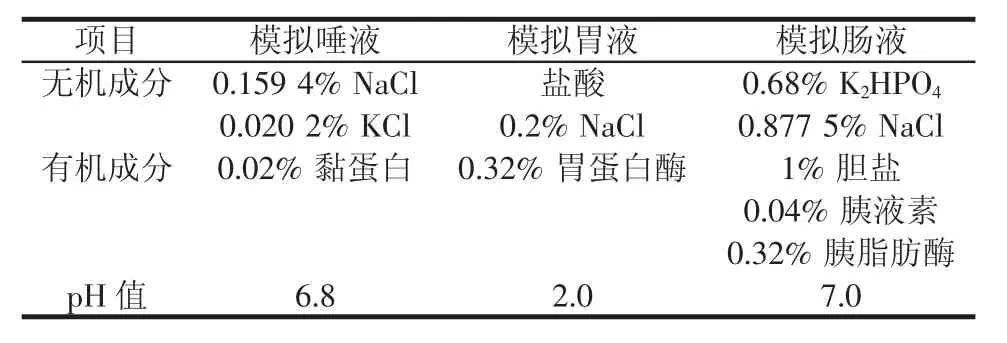
表1 模拟体外消化的消化液成分Table 1 Compositions of digestive juices in the simulated in vitro digestion
模拟唾液消化:取1 g LO/EC微胶囊于锥形瓶中,加入20mL模拟唾液,在37℃的水浴摇床中放置10min,取5 mL样品记为AS-10。
模拟胃液消化:向口腔消化后的产物中加入模拟胃液,将pH值调到2,于37℃水浴摇床中放置120 min,每隔30 min取样,立即放到冰水浴中终止反应。取出的样品依次标记为 SGF-30、SGF-60、SGF-90、SGF-120。
模拟肠液消化:向胃部消化后的产物中加入模拟肠液,其他操作步骤同模拟胃液消化过程。每次取出的样品依次标记为 SIF-30、SIF-60、SIF-90、SIF-120。
在模拟体外消化的过程中,取部分样品干燥后通过扫描电镜观察其微观结构,方法同1.3.2.2。
将不同消化阶段和消化时间后的样品稀释10倍,记录消化过程中0.01 mol/L NaOH溶液的消耗体积,计算游离脂肪酸含量,公式如下[14]。
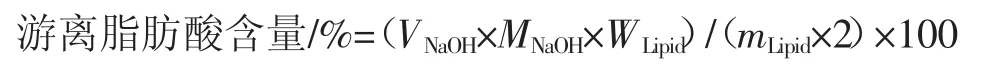
式中:VNaOH为消化过程中消耗的NaOH溶液的体积,L;MNaOH为 NaOH 的摩尔浓度,mol/L;WLipid为亚麻籽油的摩尔质量,870 g/mol;mLipid为体系中油脂质量,g。
1.4 数据分析方法
每个样品重复3组,结果用平均值±标准差表示。数据运用SPSS 18.0软件进行统计分析,通过单因素方差分析(ANOVA)、邓肯检验(Duncan)进行差异分析,显著水平为p<0.05。
2 结果与分析
2.1 壁材组成对LO/EC微胶囊性质的影响
LO/EC微胶囊的结构形态影响微胶囊的质量及芯材的释放。壁材组成对LO/EC微胶囊外观和微观结构的影响见图1。

图1 LO/EC微胶囊的外观图和微观形貌Fig.1 The morphology and microstructure of LO/EC microcapsules
图1A显示,溶液上下分层,其中上层为醇-水溶液,下层为LO/EC微胶囊。观察发现,EC-P、EC-G、EC-SP上层为不透明的醇-水混合液,表明EC颗粒在油水界面的吸附已经达到饱和。而EC-TW体系中上层为乳状液。
使用扫描电子显微镜观察LO/EC微胶囊的微观形貌,发现所有LO/EC微胶囊均呈现不规则的形态。微胶囊的表面形态取决于芯材的性质和壁材的形成过程,最初形成的油滴倾向于迅速聚结,直到它们的表面被壁材包覆。由于相邻液滴之间共享吸附的颗粒,因此体系中可能出现桥联絮凝等现象[15]。低黏度的EC分子空间排斥力较弱,静电斥力低,容易聚集成多核微粒。因此EC-P存在聚集现象,颗粒之间有相互连接和融合的趋势。此外,EC-乙醇溶液的内部凝聚力强,少量残留的溶剂会形成黏性膜,导致微胶囊间的聚结[16-17]。由图1B可知,添加剂可以降低亚麻籽油和EC之间的界面能,减少EC在固化成核阶段的聚结,并改善LO/EC微胶囊的形貌。由于G的空间位阻作用可以减小油滴尺寸和微胶囊聚集程度[18],EC-G的聚集程度减小,但是表面仍存在一定数量的小油滴。EC-SP聚集程度最小,其表面平滑、无油滴,这是因为SP可以提高聚合物链的柔韧性,降低EC间的内聚力,从而减少EC颗粒间的聚集,形成表面更加光滑的LO/EC微胶囊。ECTW与EC-P呈现相似的外观形态和明显的聚结趋势。
粒径大小和粒径分布是判断微胶囊形态的重要指标。壁材组成对LO/EC微胶囊的粒径大小和粒径分布的影响见图2。

图2 LO/EC微胶囊的粒径大小和粒径分布Fig.2 The particle size and particle size distribution of LO/EC microcapsules
由图2可知,LO/EC微胶囊粒径主要分布于180 μm~380 μm之间,并呈现单峰分布。EC-P的粒径为(374.52±1.02)μm。本试验中采用低分子量的EC原料,EC的空间排斥能较低,造成EC的生长聚集[19],因此LO/EC微胶囊的粒径相对较大。添加剂可以增加连续相的动力黏度,减小界面张力,从而减小微胶囊间的聚集。其中 EC-SP的粒径最小,为(181.88±1.89)μm,这是因为SP溶于亚麻籽油中,使油相中的表面张力迅速降低,有利于形成粒径较小的LO/EC微胶囊。SP的极性头部可以和EC主链发生相互作用,增加油滴间的排斥力[20]。而G虽然有界面屏蔽作用,可以减少油滴的聚集,但是其庞大的分子结构使分子间位阻作用明显[21],吸附速率较低,因此无法迅速吸附在油滴表面和在界面上形成致密的壁材。
2.2 LO/EC微胶囊的总油含量、表面油含量、包埋率
LO/EC微胶囊的总油含量、表面油含量及包埋率见图3。

图3 LO/EC微胶囊的总油含量、表面油含量和包埋率Fig.3 Total oil content,surface oil content and encapsulation efficiency of the LO/EC microcapsules
由图3可知,EC-P总油含量达到(66.30±2.96)%,与溶剂蒸发法制得的EC微胶囊[(65.9±0.6)%]接近[10]。EC-G与EC-P的总油含量无显著性差异,而EC-TW的总油含量最低。仅以EC作为壁材的EC-P表面油含量为(12.35±2.01)%,这是因为EC乳化性能较弱,在油含量过高时,相邻油滴间的空间排斥作用较弱,EC不足以完全包裹住油滴。添加剂使EC分子之间、EC分子和油分子之间表面能相近,增加界面活性,降低LO/EC微胶囊表面油含量。G作为亲水性大分子,具有一定的表面活性,有利于形成O/W乳液,促进EC分子在油-水/乙醇界面的吸附,因此EC-G总油含量较高。SP是亲油性乳化剂,EC与油之间的相互作用程度过高会在一定程度上削弱EC聚合物之间的氢键作用,因此含SP的LO/EC微胶囊总油含量较低。EC-SP表面没有油存在,是因为SP可以优先排列在EC分子的主链上,使EC在LO中充分延伸,并且还可以促进EC在LO中的溶解,增加LO和EC之间的相互作用,从而提高包埋率。SP的加入使壁材组成和油成分之间相似性增加,提高了EC-SP的包埋率。EC-TW形成后,大部分油滴存在于上层乳液中,因此EC-TW包埋率较低,为(74.93±2.31)%。此外粒径大小也是影响包埋率的主要因素,粒径小则包埋率更高[22]。本研究结果表明,使用不同类型的添加剂,LO/EC微胶囊包埋率差异显著。由于TW对改善LO/EC微胶囊形态、包埋率作用较小,后续试验针对EC-P、EC-G、EC-SP展开。
2.3 傅里叶变换红外光谱和DSC分析
傅里叶变换红外光谱可以间接判断LO是否被包埋于EC微胶囊。图4为在4 000 cm-1~500 cm-1的波长范围内EC、LO和LO/EC微胶囊的红外光谱图。

图4 EC、LO、LO/EC微胶囊的红外光谱Fig.4 FTIR spectra of EC,LO and LO/EC microcapsules
EC含有大量的羟基、醚键、甲基和亚甲基类结构,由图4可知,EC-P与LO在C-H伸缩振动(2 927、2 854 cm-1)、C=O 伸缩振动(1 745 cm-1)、C-H 弯曲振动(1 462、1 376 cm-1)、C-O 伸缩振动(1 162 cm-1)、-CH2振动(721 cm-1)的特征峰紧密匹配,表明EC包埋了LO[23]。而EC-SP的图谱显示EC在C-H拉伸的特征峰(2 974、2 928、2 871 cm-1)覆盖了 LO 的特征峰(3 010、2 926、2 854 cm-1),EC 在 C-O-C 拉伸(1 109 cm-1)的特征峰则覆盖了LO的特征峰(1162cm-1),证实LO被EC完全包埋。EC和LO均为疏水性组分,LO可能通过疏水作用与EC发生相互作用从而促进油脂的包埋[19,24]。
FTIR图谱中峰形的位移和强度变化表明分子间相互作用的发生。LO/EC微胶囊中没有出现新的特征峰,表明EC包覆LO过程中未产生新化合物。与EC相比,EC-P和EC-G在3 472 cm-1处(O-H拉伸)特征峰强度降低,说明EC分子间的氢键作用力减弱。与EC-P相比,EC-G中的特征峰由3 010 cm-1迁移至3 011 cm-1,2 854 cm-1迁移至 2 855 cm-1,C-H 键的微弱迁移可能是因为G促进EC与LO之间的疏水作用。EC-SP在3 472 cm-1的O-H拉伸强度增加,是因为SP的极性基团促进EC的羟基暴露,有助于EC分子间结合,并且促进EC主链和LO的相互作用,增强EC和LO的羟基之间的氢键作用。
表2总结了样品的玻璃化转变温度(glass transition temperature,Tg)、峰值温度(peak temperature,Tp)和焓变值(ΔH)。

表2 EC及LO/EC微胶囊的DSC图的温度及焓变值Table 2 Temperature and enthalpy change of DSC diagram of EC and LO/EC microcapsules
由表2可得,与EC-P相比,EC-G和EC-SP的玻璃化转变温度升高,焓变值增加,说明热稳定性提高。LO/EC微胶囊的DSC热力学性质变化可分为3个阶段:第一阶段在100℃左右,EC分子和LO/EC微胶囊在100℃左右时存在微小的吸热峰,这可能与聚合物中结合水的蒸发有关[25]。研究发现吸热峰位移与水-聚合物相互作用的强度以及持水量有关[26],EC-G和ECSP的焓变值更高,对水的亲和力增强,表明产品热稳定增强。第二阶段是玻璃化转变阶段,聚合物的无定形区域获得能量而摆脱强键束缚,由玻璃态转变为橡胶态[27-29]。本试验黏度为10 cp的EC分子量低,EC基线在100℃附近开始变化,Tg为(105.24±2.27)℃。EC-G、EC-SP 的Tg显著提高,分别为(112.61±2.09)、(114.75±2.81)℃。第三阶段的Tp和ΔH表示当物质由有序的结构全部转变成无序的结构时的温度和所需要的能量[30]。此时LO与EC的分子间作用力被破坏,壁材成分开始分解。相比于EC,EC-P表面油含量较高,其峰值温度则更低。而EC-G和EC-SP的峰值温度和焓变值均高于EC-P,表明LO/EC微胶囊的热稳定性提高[31]。
2.4 贮藏稳定性分析
37℃贮藏过程中LO和LO/EC微胶囊的过氧化值变化见图5。
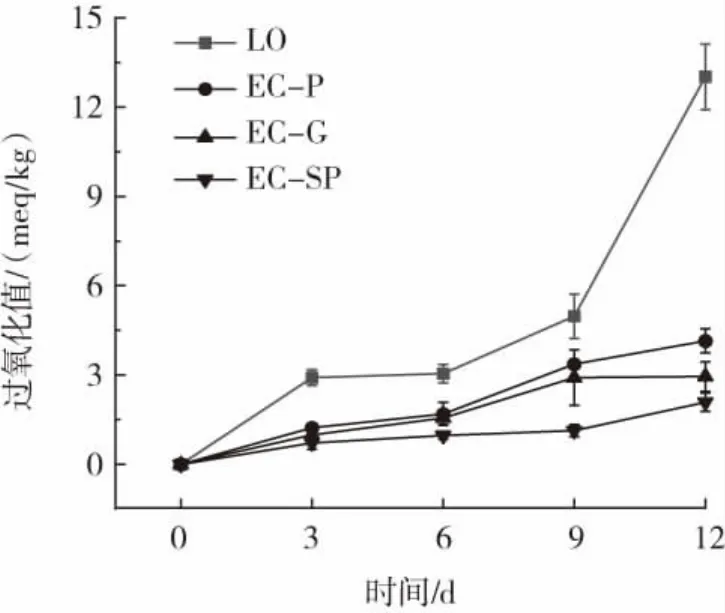
图5 LO和LO/EC微胶囊在37℃贮藏过程中过氧化值变化Fig.5 Peroxide value of linseed oil and LO/EC microcapsules at 37℃storage
通过测定过氧化值来评估LO/EC微胶囊在12 d(37℃)贮藏期间的氧化稳定性。由图5可知,所有样品的过氧化值在最初3 d内快速增加,这是由于LO/EC微胶囊制备中的加热过程导致的。随着时间的延长,LO与LO/EC微胶囊的过氧化值继续增加。与亚麻籽油相比,LO/EC微胶囊的氧化程度降低。首先,表面油的存在会加速氧化的过程,随着表面油含量的减少,其氧化稳定性明显提高,其次,壳层厚度增加也有助于提高氧化稳定性。在微胶囊的贮藏过程中,稳定的界面屏障可以明显减少氧气、金属离子等与油脂之间的相互作用,从而提高油脂的氧化稳定性。因此LO/EC微胶囊比亚麻籽油具有更高的氧化稳定性。在37℃贮藏12 d,LO/EC微胶囊的过氧化值仍符合国家标准中对新鲜油脂过氧化值的要求(<10 meq/kg)。
2.5 模拟体外消化结果
LO和LO/EC微胶囊在消化过程中的脂肪酸释放率见图6。

图6 LO和LO/EC微胶囊在消化过程中的变化Fig.6 The change of LO and LO/EC microcapsules during digestion
图6表明,LO/EC微胶囊在唾液和胃液中的消化程度低,不足以破坏微胶囊的壁材。经过AS和SGF消化后,LO的释放率较低[(5.52±1.12)%~(11.59±1.01)%]。SGF中的消化主要表现为表面油的分解,而在SIF消化过程中,微胶囊壁材结构开始被破坏,因此脂肪酸在SIF消化阶段大量释放。在SIF消化阶段的前30 min,LO及LO/EC微胶囊的脂肪酸释放率迅速提高[(23.28±0.80)%~(46.60±2.10)%]。界面的厚度、界面层的位移等因素均会影响油脂水解速率[32]。各样品中的脂肪酸最终释放程度为LO>EC-P>EC-G>ECSP。在SIF中消化120 min后,EC-P、EC-G、EC-SP脂肪酸释放率均低于LO。EC不易消化,可以形成稳定的物理屏障,阻止油和消化液的接触,延缓油脂在胃肠液中的消化。尽管EC-G和EC-SP体积更小,与消化液的接触面积更大,但是G和SP可以加强EC与LO之间的相互作用,使壁材更为坚固,导致其消化更为缓慢。随着消化时间的延长,LO/EC微胶囊仍持续缓慢地释放脂肪酸。LO/EC微胶囊在消化过程中的微观结构见图7。图7显示,EC-P和EC-SP在消化过程中结构变化存在差异。EC-P经SGF消化120 min后,开始少量聚集。EC颗粒(壁材)在pH2时通常会发生聚集,胃液中的强酸环境使界面静电屏蔽作用降低,诱导EC壁材间发生桥联絮凝作用[33]。此外表面油消化完全后,会促进絮凝作用。同时胃酸和胃蛋白酶对微胶囊的表面有侵蚀作用,使微胶囊结构不稳定而发生聚集。在SIF消化120 min后,LO/EC微胶囊被胰液素和胆盐消化而溶蚀崩解,EC-P形成表面粗糙的大尺寸聚集体。与EC-P相比,EC-SP受胃肠液消化的影响相对较小。在强酸性胃液消化120 min后,EC-SP并未发生团聚。相比在肠液消化后,在SIF消化120 min后EC-SP开始聚集,但仍基本保持原状。

图7 LO/EC微胶囊消化过程中的微观结构Fig.7 Microstructure of LO/EC microcapsules during digestion
3 结论
本研究采用反溶剂法制备LO/EC微胶囊,在EC颗粒形成的同时包埋亚麻籽油,方法简单,油脂包埋效率高。研究显示,壁材组成会影响LO/EC微胶囊的壁材结构和油脂包埋率,SP的加入可以增强EC和亚麻籽油之间的氢键作用和疏水作用,使LO/EC微胶囊具有最高的油脂包埋率,并显著提高微胶囊的热稳定性、氧化稳定性,延缓油脂消化速率。G的加入会促进EC和亚麻籽油之间的疏水作用,改善LO/EC微胶囊及其包埋油脂的稳定性,并使LO/EC微胶囊保持较高的总含油量。但是LO/EC微胶囊存在容易聚集的问题。未来可以深入研究EC颗粒作为壁材形成微胶囊的作用机理以及EC颗粒固化成壁层的影响因素,以改善其稳定性。
猜你喜欢
食品工业科技(2022年21期)2022-10-27
农产品加工(2022年14期)2022-08-18
科学导报·学术(2020年89期)2020-12-08
食品界(2019年8期)2019-10-07
山东农业科学(2018年8期)2018-11-22
中国纺织(2017年12期)2017-12-20
散文诗世界(2017年3期)2017-11-13
家庭百事通·健康一点通(2016年7期)2016-08-04
绿色科技(2016年7期)2016-05-14
食品与生活(2014年4期)2014-05-27
